In the first reported correlation between caeruloplasmin and cardiovascular disease in 1956, Adelstein et al. showed increases in serum caeruloplasmin after patients suffered heart attacks. Since that report, elevated serum caeruloplasmin levels have been found in patients with a multitude of cardiovascular disorders including atherosclerosis, abdominal aortic aneurysms, angina and peripheral arterial disease. While these correlations may partly be explained by the involvement of caeruloplasmin as an acute phase reactant, 1 known to participate in the inflammatory response to these pathologies, several studies have indicated that caeruloplasmin, or copper, may be an independent risk factor for cardiovascular disease. Studies on the same cohort showed that an elevated serum level of LDL was associated with accelerated atherosclerosis only in subjects with higher than median serum copper levels.
The possibility that serum caeruloplasmin, and so copper, level is an independent risk factor for coronary heart disease remains to be clarified by further investigation into the multifaceted chemical mechanisms concerned.
Coronary artery disease (CAD) is the development of atheromatous plaques which harden and constrict the walls of the arteries feeding the heart. The burden of CAD is greatest in the Western hemisphere, where symptomatic angina and heart attacks are widespread and frequently fatal. As well as accounting for approximately 3% of all hospital admissions; CAD kills more than 110,000 people a year in England, of who more than 41,000 are under the age of 75.
The principal treatment for advanced CAD is coronary artery bypass graft surgery (CABG). Despite the impressive short-term success of the technique, as many as 50 % of grafts fail within 10 years leaving the search for a therapy effective in reducing vein graft failure a matter of urgency2. It is imperative to further knowledge of the mechanisms fundamental to the disorder, particularly the impact of suspected risk factors and oxidant stress, which are areas currently under-investigated.
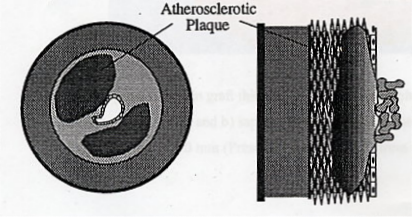
The artery is comprised of three basic layers; the intima, the media and the adventitia. The formation of an atheromatous plaque involves a dramatic morphological change within the artery which results in a gross enlargement of the intima, a concomitant reduction in lumen diameter and is often accompanied by wasting of the media. When this atherogenesis occurs in the coronary artery it results in a number of debilitating or life-threatening disorders, including angina pectoris, myocardial infarction and heart failure.
Atherosclerosis is a progressive disease which is initiated by the deposition of cholesterol within the arterial wall. Such lipid deposits are known as "fatty streaks" and progress to small but distinct lesions, called neointima, upon which the atheromatous plaque may then begin to develop.3 The continual deposition of cholesterol, along with the matrix proteins collagen and elastin, results in a plaque with a characteristic opaque-white appearance and an infiltration of microvessels from adjacent vascular tissue. In time the plaque may develop to a degree that elicits clinical symptoms; for example, the narrowing of the arterial lumen results in angina pectoris, acute chest pain caused by the insufficient oxygenation of the heart muscle tissue. More seriously, the advanced plaque can break away and cause a thrombosis, blocking a coronary artery and killing heart tissue, so causing a "heart attack".
Superimposition of atheromatous plaques leads to significant narrowing of the artery.
The formation of atheromatous plaques is accelerated in patients with other clinical conditions; principally diabetes mellitus, hypertension and obesity, whilst lifestyle factors such as tobacco and alcohol addiction also facilitate the inception of CAD.75
Although there is no pharmacological therapy that can prevent plaque rupture, there are several available drugs which can retard the progress of atherogenesis. However, in the occurrence of an advanced coronary artery plaque, patients must be treated by surgical intervention, principally CABG. CABG restores myocardial blood flow and reduces the risk of mortality due to occlusive thrombosis. It is one of the most commonly performed surgical procedures in the Western World, with more than 40,000 such operations being performed annually in the UK and more than 400,000 in the USA.
The procedure involves grafting bypasses into the aorta and coronary artery using sections of the saphenous vein, previously removed from the patients` lower leg. The bypasses are positioned downstream of the atherosclerotic lesion such that they circumvent the plaque and hence restore adequate blood flow to the myocardium.
Despite its widespread and effective use, the Achilles heel of CABG lies in the subsequent progressive decline in graft patency over time. Early vein graft failure due to thrombosis either results from trauma during surgical preparation or anastomosis to a small native coronary artery with an inadequate distil "run-off". Thrombosis is responsible for the failure of 8 - 18 % of vein grafts in the month following surgery. Grafts which do survive this early period go on to exhibit a progressive graft thickening as medial vascular smooth muscle cells (VSMCs) proliferate and migrate to the intima. This is the direct cause of the increased lipid content observed in the graft wall between the first and fourth years after implantation and is hence the principal precursor event in atheroma formation.74, 75
Obvious atherosclerotic changes in vein grafts are first observed at three years and then seen to accelerate beyond five years with the appearance of mature lipid-laden atherosclerotic plaques. Ten years after surgery less than 50 % of grafts are patent, with the remainder showing angiographic evidence of significant vein graft disease. While these lesions essentially have the same cellular composition as arterial lesions, they tend to be more diffuse and friable, thus predisposing them to plaque rupture and thrombosis.4
The corrective operation and subsequent medical care following vein graft failure costs approximately £12,000. Considering 20,000 patients require this treatment every year in the UK, devising novel strategies to reduce the incidence of late vein graft failure is an area of considerable importance at both an economic and a human level.
The development of effective strategies to prevent vein graft failure has been an area of intensive research. The administration of anti-thrombotic drugs, in particular aspirin, has reduced the incidence of early vein graft failure due to thrombosis, although this treatment is less than optimal. In the case of late vein graft failure, only aggressive lipid lowering therapy has proved successful and this only worked in cases of hypercholesterolaemia.
The general consensus is that the inhibition of neointima precursor formation will inhibit the formation of superimposed atherogenesis and concomitantly late vein graft failure. In turn, devising effective interventions requires a full understanding of the pathological processes underlying neointima formation, and especially the factors which promote oxidant stress.
The risk factors for late vein graft failure are essentially the same as those for CAD; the most important being diabetes mellitus, hypercholesterolaemia and hyperhomocysteinaemia, which leads to oxidant stress.
There is an extremely strong positive correlation between patients with raised cholesterol levels and those suffering vein graft failure. The grafted saphenous vein has high lipid permeability, which in conjunction with high pressure and hypercholesterolaemia, leads to enhanced lipid uptake and retention in vein grafts. The mechanism underlying this process is associated with the release of chemoattractants and growth factors which stimulate the smooth muscle cell migration and proliferation, propagating the formation of the atheromatous plaques which prompt vein graft failure.74, 76
Diabetes mellitus also increases the mortality rate in patients who have undergone CABG. The mechanism behind the interaction of this risk factor has not been widely investigated although it has been firmly established that both diabetes mellitus and hypercholesterolaemia are associated with superoxide radical formation and nitric oxide (NO) reduction in the blood plasma. This is compliant with the hypothesis that O2-∙ mediated reduction of NO contributes to vein graft disease.
Furthermore, promotion of oxidant stress and the associated reduction in NO may be assigned to hyperhomocysteinaemia (HHC); patients with this condition are also exposed to a higher risk of vein graft failure.5, 6. So, is there some form of synergy between caeruloplasmin and homocysteine and possible vein graft failure?
Homocysteine (HCy) is an amino acid intermediate in a multitude of biochemical pathways and thus always present in the blood plasma. There is accumulating evidence that even a moderately raised level of Homocysteine, mild Hyperhomocysteinaemia, is associated with increased risk of CAD and vein graft failure following CABG. 7 Various genetic causes for this increase have been proposed although a variety of conditions, including dietary folic acid deficiency, may also contribute.
How HCy affects the blood vessels remains uncertain but it appears to be implicated in early changes in structure and function of the artery wall. It has been suggested that prolonged exposure to HCy can affect the way in which vessels dilate and as a consequence enhance the development of atherosclerosis. There is also evidence that moderate HHC increases the risk of thrombosis. 8
Homocysteine is a product of the reversible hydrolysis of S-adenosylhomocysteine (SAH). High concentrations of HCy are thought to lead to its auto-oxidation and the consequential generation of the two reactive oxygen species, hydrogen peroxide (H2O2) and superoxide radicals (O2-∙). 9
Oxidant stress is a condition in which cells are exposed to excessive levels of either molecular oxygen or reactive oxygen species. Principal among these reactive oxygen species is superoxide (O2-∙) and hydrogen peroxide (H2O2) whilst other oxidants include hyperchlorous acid (HOCl), hydroxyl radicals (OH∙), reactive aldehydes, lipid peroxides and lipid radicals.
There are many potential biochemical sources of reactive oxygen species including the mitochondrial electron transport chain, the redox cycling of small molecules, auto-oxidation and hyperoxic conditions, although studies have established that the major enzymatic source of O2-∙ in blood vessels is NADPH oxidase, as expressed by VSMCs.
Despite the fact all tissues possess mechanisms designed to negate the impact of reactive oxygen species they are likely to be subjected to a certain degree of oxidant stress. This may adversely affect vein grafts at both a blood and tissue level where the initial adhesion of platelets and leukocytes generates the O2-∙ which elicits acute local intra-graft effects; such promotion of inflammatory cascades and vasoconstriction may in turn lead to graft failure.
Considerable evidence indicates that OS is central to the pathological impact of HCy. The amino acid is known to auto-oxidise to form a mixture of disulphides including its corresponding dimer homocystine. 10 The reaction involves significant generation of H2O2 and O2-∙ which lead to the, afore mentioned, proatherogenic affects.
The results of recent studies have demonstrated that, following CABG, there is a significant increase in plasma HCy, copper and caeruloplasmin that manifests itself within a week of operation and is maintained for at least 6 weeks. Considering the initiation of neointima formation occurs within this time scale, the data may be of considerable importance in the rationalisation of the pathophysiology and treatment of late vein graft disease. Firstly, the significant increase in HCy may contribute to vein graft thickening through the mechanism of oxidant stress, which is central to its pathological impact. 11, 12, 13, 14 A number of factors are known to promote an increase in plasma HCy, most notably a decrease in blood folate and vitamin B12 levels, although, in the recent study, these remained unchanged in the 6 weeks following CABG and were not below the normal levels pre-operatively.
The strong correlation between plasma copper and serum caeruloplasmin concentrations reflects the fact that the majority of plasma copper exists bound to caeruloplasmin.15 It has long been recognised that surgery can increase blood copper levels, 16 although the discovery that blood copper levels were significantly correlated with those of HCy may be of profound relevance to the initiation and augmentation of vein graft thickening.
Correlation has also been made between the level of blood plasma copper and generation of superoxide radicals, O2-∙. It is proposed that the presence of copper may perceptibly increase the potency of HCy whilst elevated total plasma copper levels have been observed in patients with hyperhomocysteinaemia. It is proposed that the concomitant increase in HCy and plasma copper may play an aetiological role in vein graft disease.
If it is not the absolute concentration of plasma HCy, but the relative concentrations of copper (and caeruloplasmin) that are of pathological significance, then the administration of antioxidants or superoxide dismutase mimetics may prove beneficial to future CABG patients.
The "in vitro" behaviour of caeruloplasmin and homocysteine was examined during the MSc Final Year Undergraduate research project by Caroline Green at Bristol, and preliminary evidence was obtained that homocysteine is oxidised to its disulphide form by caeruloplasmin.17
1. Rice, E.W., Clin. Chim. Acta. (1961) 6, 625-655.
2. Mortwani, J.G., Topol, E.J., Circulation(1998) 97 916 - 931.
3. Jeremy, J.Y., Mehta, D., Bryan, A.J. et.al.,Platelets, (1997) 8, 295 - 309.
4. Shukla, N., PhD thesis, University of London (2001).
5 Boushey, C.J., Beresford, S.A.A., Omenn, G.S. and Motulsky, A.G., JAMA, (1995), 274, 1049 - 1057.
6 Graham, I.M., Daly, L.E., Refsum, H.M. et.al., JAMA, (1997) 277,1775 - 1781.
7. Refsum, H., Ueland, P.M., Nygard, O. and Vollset, S.E., Caeruloplasmin, Ann. Rev. Med. (1998) 49,31- 62.
8. Chambers, J.C., Seddon, M.D. and Shah, S. , Roy. Soc. Med. (2001) 94 10-13
9. Loscalo, J., J. Clin. Invest, (1996) 98 5 -7
10. Chinmay, K., Mukhopadhyay, L. and Fox, P.L. ,Biochemistry (1998) 37, 14222 - 14229
11. Starkebaum, G. and Harlan, J.M. ,J. Clin. Invest, (1986) 77 1370 - 76
12. Emsley, A.M, Jeremy, J.Y., Gomes, G.N. et. al. ,J. Pharmacol (1999) 126, 1034 -1040
13 Emsley, A.M., Plane, F., Jackson, C. and Jeremy, J.Y. ,Vascular Disease ,(1998) 1, 3 - 8
14. Jeremy, J.Y., Rowe, D., Emsley, A.M. and Newby, A.C. , Cardiovas. Res. ,(1999), 43 658 - 665
15. Antila, H., Salo, M., Nato, V. et.al. J. Parent. Ent. Nutr.,(1990) 14, 85 -89
16 Hallbook, T. and Hedlin, H. (1980) 146 371-373.
17 Green, C.M.H., (2003), MSci Thesis, School of Chemistry, Bristol University. (Unpublished)