Erasmus MC-University Medical Center, Rotterdam, Holland
[This webpage was written as part of the Molecule of the Month collection for Sept 2003]

L-carnitine, systematic name (R)-(3-Carboxy-2-hydroxypropyl) trimethyl-ammonium hydroxide, is soluble in water and ethanol. In aqueous solution at a physiological pH it is a buffer. Carnitine is present in all animals, in bacteria and in some plants. In our body it has been detected in all cells and body fluids in highly variable concentrations. Most carnitine is stored in muscle. Carnitine acts as a carrier of activated fatty acids of variable chain length, esterified to its hydroxyl group. The name of these esters is acylcarnitine. Carnitine is indispensable for the mitochondrial beta oxidation of acyl-CoA esters, because it is transported with the acyl moiety, over the mitochondrial inner membrane. The same machinery is also used for export of acetyl and other short-chain acyl groups out of the mitochondria. The carnitine system is present in all cell types, even in cells without mitochondrial fatty acid oxidation such as neurons and granulocytes and even in red blood cells, a cell type without mitochondria. Carnitine is a vitamin for babies who consume it with milk. It is not a vitamin for healthy adults [1-4].
Carnitine as pdb file (open with Rasmol).
1905 Carnitine was discovered in meat extract.
1927 The correct structure of carnitine was proposed.
1948 A new vitamin BT was discovered, essential for growth of the yellow mealworm. The T was derived from the Latin name of this species, Tenebrio mollitor.
1952 Vitamin BT was identified as carnitine.
1955 Discovery of the enzyme carnitine acetyltransferase (CRAT).
1958 I.B. Fritz discovered that carnitine stimulates long-chain fatty acid oxidation by mitochondria.
1963 Discovery of the enzyme carnitine palmitoyltransferase (CPT)
1973 First papers about patients with inborn errors in carnitine metabolism: muscular carnitine deficiency and CPT deficiency.
1975 Discovery of carnitine acylcarnitine translocase (CACT).
1977 Discovery of the inhibition of CPT-I by malonyl-CoA.
1977 Discovery of CPT-II deficiency with normal CPT-I, and the presence of CPT-I and not CPT-II in erythrocytes.
1982 Discovery of a carnitine transport defect as the cause of primary systemic carnitine deficiency.
1987 Discovery of the localization of CPT-I in the outer mitochondrial membrane.
1988-today Gene analysis of enzymes and transporters of the carnitine system in humans, patients with inborn errors and (transgene) animals. This biochemistry at the DNA-level has not been reviewed here.
1991 Discovery of fatal CPT-II deficiency.
1992 Discovery of fatal carnitine acylcarnitine translocase deficiency.
The carnitine system consists of L-carnitine and acylcarnitine esters and the enzymes and transport proteins required for their metabolism and transport, including carnitine acyltransferases, mitochondrial carnitine acylcarnitine translocases, plasma membrane carnitine importers, and the carnitine biosynthesis pathway from lysine and methionine. In contrast to humans, bacteria possess enzymes to catabolize carnitine. Intestinal bacteria degrade most of the orally supplemented carnitine [2].
1. Acylcarnitines
Acylcarnitine esters are formed from the CoASH esters of acetate, propionate, butyrate, medium-chain, long-chain and very-long-chain fatty acids. The highest possible chain length communicating with the carnitine system is not precisely known, but it is likely, although not proven with certainty, that physiological important fatty acids such as arachidonic acid (C20:5, n-6) and docosahexaenoic acid (C22:6, n-3) are transported as acylcarnitines to the membranes where they are needed. In blood reside carnitine esters up to C18, but in the tissues higher acylcarnitines were encountered [6]. Also most substituted-, branched-chain- and dicarboxylic acyl-CoA esters are converted into acylcarnitines, but at a (much) slower rate than the straight-chain esters.
2. Carnitine acyltransferases
The family of enzymes responsible for the reversible acylcarnitine formation are named carnitine acyltransferases and they catalyze the following reversible reaction.
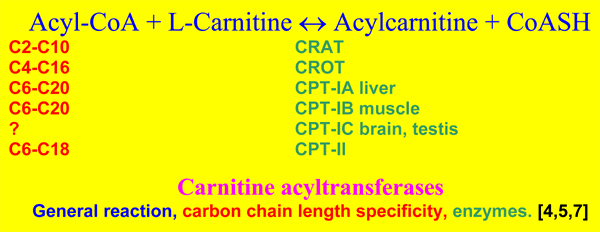
The carnitine system was first discovered in mitochondria. They were found to contain carnitine acetyltransferase (CRAT), firmly bound to the inner face of the inner mitochondrial membrane and four CPT isoenzymes, named CPT-IA in liver, CPT-IB muscle and most other cells, CPT-IC in brain and testis [7] and CPT-II. The CPT-I enzymes are all localized in the outer mitochondrial membrane, with their catalytic [8]and regulatory [9,10] domains for malonyl-CoA inhibition facing to the cytosolic side, while CPT II is localized as CRAT. Peroxisomes contain also CRAT, a membrane carnitine octanoyltransferase (CROT) inhibited by malonyl-CoA, and another soluble carnitine acyltransferase not inhibited by malonyl-CoA[4]. So it is clear that ac(et)yl transport is not confined to the mitochondria. Also endo- and sarcoplasmatic reticulum, the nuclear membrane and the plasma membrane contain carnitine acyltransferases [4].
3. CACT
Carnitine acylcarnitine translocases reside in the mitochondrial inner membrane and catalyze the import of acylcarnitine of variable chain lengths usually in exchange for carnitine or short-chain acylcarnitine. Brain and testes mitochondria possess another CACT, which is also induced in liver after partial hepatectomy [11].
A third mitochondrial CACT transports branched-chain acylcarnitine molecules [12]. In this respect it is of interest that peroxisomal CROT is involved in branched-chain fatty acid metabolism[13].
4. Carnitine importers
The high affinity transporter for carnitine is named OCTN2, a neutral organic cation transporter [14-16], which is stimulated by NaCl [2,17]. This transporter is not fully specific for carnitine, but transports also other drugs. OCTN2 is localized in the plasma membrane of muscle, heart, and the brushborders of small intestine [18] and kidney, and many more cells. OCTN3 had an intermediate affinity for carnitine, is not stimulated by Na+ and predominantly expressed in the testis [15]. OCNT1 has such a low affinity for carnitine that it cannot act at physiological carnitine levels [19].
CT2, carnitine transporter 2, a novel carnitine transporter, is expressed at the luminal membrane of the epididymal epithelium [20]. OCTN2 is also present in these cells, but in the basolateral membrane [21]. The carnitine transporter in the basolateral membranes of the mouse kidney and small intestine, has also been characterized. It is another high affinity transporter, not stimulated by Na+ [22].
5. Biosynthesis
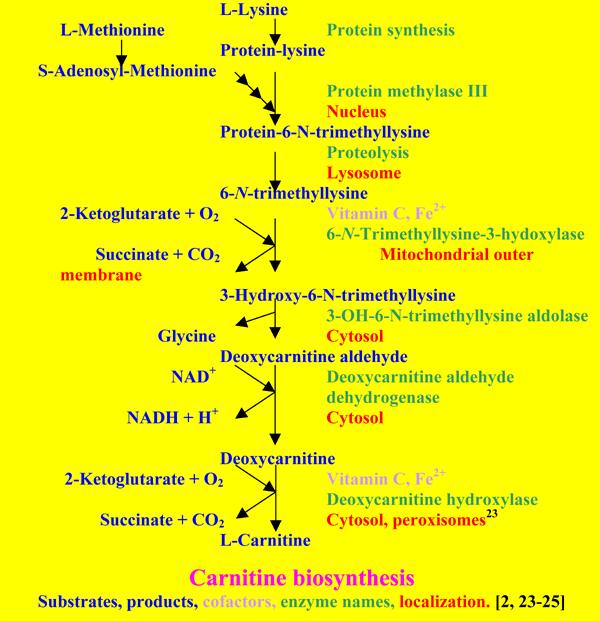
Most tissues are able to produce deoxycarnitine [24, 25], but the last step is in humans only present in liver, kidney, brain and testes. Only liver and kidney are able to export carnitine to the blood. In healthy adults this biosynthetic pathway may provide enough carnitine to compensate for the urinary loss. In babies it is not enough to meet the demands of the growing body [2, 24]. The carnitine system is regulated via peroxisome proliferator-activated receptors [26] and hormones such as insulin, glucagon, norepinephrine, thyroid hormone, dopamine and androgens [3].
1.-Carnitine is indispensable for the mitochondrial oxidation of long-chain fatty acids.
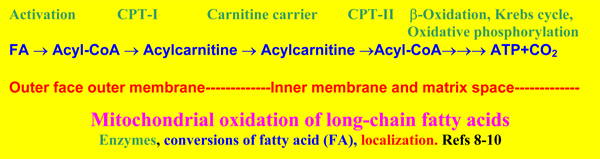
Long-chain fatty acids that are used as energy source, are activated by the palmitoyl-CoA synthetase localized on the outer surface of the outer mitochondrial membrane [27, 28], and converted by CPT-I at the same site [8-10].
There are many acyl-CoA synthetases, differing in acyl-specificity from acetyl-CoA to lignoceroyl-CoA, and in localization [29-32]. Acyl-CoA synthetases are the gateway to mitochondrial or peroxisomal �-oxidation, or synthesis of triacylglycerols, phospholipids and other acylated products, while the CoA esters and acylated proteins are also acting in cell signaling [33].

By the subsequent action of CPT-I on the outer surface of the outer mitochondrial membrane, the carnitine carrier and CPT-II on the inner surface of the inner mitochondrial membrane, palmitoyl-CoA virtually is transported into the
matrix, but note that its CoASH comes from the matrix space, while initially the CoASH is from the cytosol. Palmitoyl-CoA is then oxidized by the enzymes of the mitochondrial �-oxidation and the oxidative phosphorylation, to synthetize energy in the form of ATP, the ultimate purpose of these reactions.
The mitochondrial �-
oxidation spirals are also catalyzed by enzymes of different acyl-specificity, with extra enzymes for the degradation of unsaturated fatty acids [34-35]. The acetyl-CoA produced is oxidized to CO2 and energy (plus free CoA) in the Krebs cycle of all mitochondria. It may be temporarily converted into acetylcarnitine, and in the fasting liver in ketone bodies. These molecules are exported to the blood, and are oxidized via acetyl-CoA in other cells.
In other organs than the liver, the mitochondrial oxidation of fatty acids is firmly linked to the Krebs cycle.
It is generally thought that medium-chain fatty acids do not need carnitine for transport to the matrix. That is true for the liver, but not for (rat) muscle and heart [36]. Another believe is that most carnitine is needed to enable acyl-transport, but experiments with rat muscle mitochondria revealed that the bulk of carnitine was needed to liberate CoASH from the produced acetyl-CoA, because carnitine could be replaced by L-malate [3].
2.-Carnitine stimulates mitochondrial metabolism by decreasing acyl-CoA and increasing CoASH. Removal of long-chain acyl-CoA de-inhibits adenine nucleotide translocator, glutamate dehydrogenase, CRAT etc. Removal of acetyl-CoA de-inhibits pyruvate dehydrogenase, which leads to decreased lactate levels and increased glucose oxidation [3]. Removal of branched-chain acyl-CoA in muscle de-inhibit branched-chain acyl-CoA dehydrogenases. Branched-chain-acylcarnitines are further oxidized in liver [37].
3.-Carnitine decreases oxidative stress and inhibits apoptosis by preventing ceramide formation from palmitate [38] and opening of the permeability transition pore [39]. It inhibits caspases [40].
4.-Carnitine and also acetylcarnitine protects against poisoning by (neuro)toxins [41-45] and corrects mitochondrial, lipid and neurotransmitter receptor abnormalities in aging brain and heart [46-50].
5.-Carnitine stabilizes membranes, abolishes Ca2+-overload and stimulates the microcirculation in ischemia [51-54].
6.-Carnitine is involved in membrane repair by reacylation, a process occurring in all cells from erythrocytes to neurons [55,56].
7.-Carnitine restores erythrocyte fluidity, enabling passage through capillaries [57]
8.-Carnitine stimulates the immune response and decreases inflammatory cytokines [58-62].
9.-Carnitine promotes maturation of the fetus [63], lungs [64] and sperm [65-66].
10.-Carnitine is involved in acetyl-delivery to choline, aspartate, lipids, histones, and proteins. Acetylation is an important post-translational modification of proteins.
11.-Carnitine is involved in membrane acylation and G-protein signaling [67].
12.-Carnitine transforms, with little electrical stimulation, fibre type II to I in dog muscle [68].
This is a selection of the manifold number of functions and actions of carnitine and its system. Entering the era of genomics, proteomics and lipidomics, it will be clear that the complete story is not yet told.

The defects in the carnitine acyltransferases found until now are confined to the mitochondria. As with several disorders in mitochondrial (very-) long- chain fatty acid oxidation, the mild form of CPT-II affect mainly muscle, with exercise-induced myoglobinuria as presenting symptom. The severe forms of CPT-II and CACT defects are fatal multi-organ disorders with cardiomyopathy and low carnitine levels in tissues and blood. CPT-I liver deficiency affects liver and causes a defective hepatic production of glucose and ketone bodies. As may be expected blood carnitine is not decreased [35].
OCTN2-deficiency is the same as primary systemic carnitine deficiency and is discussed in the next section about carnitine deficiency.
Defects in OCTN2 causes primary systemic carnitine deficiency presenting with cardiomyopathy and often myopathy and/or hepatic encephalopathy in early childhood, and is a treatable condition. These patients are cured by carnitine supplementation. Before treatment, the carnitine levels in muscle and blood are very low [68-70].
Secondary carnitine deficiency results from three main, sometimes overlapping, causes.
Muscle carnitine is also deficient in disuse, muscle atrophy by innervation problems, muscular dystrophy, mitochondrial myopathy and other myopathies, and after ischemia and reperfusion [69]. Muscle carnitine transporter deficiency has been reported [5].
There are many assays for carnitine. Here follows a selection of 4 commonly used methods.


Carnitine has been supplemented in a great variety of patients, such as inborn errors in the carnitine system, mitochondrial beta oxidation, mitochondrial acyl-CoA-li
nked enzymes, oxidative phosphorylation [35,84], in cardiovascular diseases [85], AIDS [60], in diabetes mellitus [86], chronic fatigue syndrome [87], chemotherapy- [88] and interferon-a- [89] induced fatigue, children with ADHD [90]. Not only carnitine itself, but also small carnitine esters are used for therapy. Acetylcarnitine for brain diseases [91-93] propionylcarnitine for cardiovascular disorders [85]. Both compounds were found to be about equally effective in an open study of chronic fatigue syndrome patients, but acetylcarnitine had most effect on brain fatigue and propionylcarnitine on general fatigue [94]. Plasma levels of carnitine in these patients, and also in the earlier mentioned ADHD patients [90] were normal.Carnitine supplementation is also advocated for healthy babies who are not consuming milk, pregnant and lactating women, for sporters, in malnutrition and in animal feeding.
Carnitine, acetylcarnitine and propionyl-carnitine are safe and have only few side effects in a minority of the patients, such as diarrhea, sleeplessness [94] or trimethylamine formation [95], the odor of rotten fish in skin and hair. This usually disappears by taking less carnitine, or by reducing the activity of the intestinal bacteria [95] or by supplementation with extra (10 mg/d) riboflavine [96]. Hundreds of patients were treated with acetylcarnitine and propionyl-carnitine, but trimethylamine formation was not observed [97].
There is general agreement that carnitine supplementation may be life saving in a variety of conditions. For instance carnitine normalizes heart size and function in children with primary carnitine deficiency [69, 70], and it prevents the neurological suffering and fatal outcome in glutaryl-CoA dehydrogenase deficiency [98]. In other conditions such as secondary deficiency due to MCAD deficiency [99] and hemodialysis [100, 101] there is less agreement in the literature. However, it is important to realize that an important number of the patients may benefit from the medication, that hepatic, renal, cardiac or neurological deterioration and sudden unexpected death could be prevented, that the life span of erythrocytes is increased [57] and the quality of life in a variety of chronic diseases is improved (e.g. ref 102).
In early days carnitine was isolated from animal muscle products and the most pure L-carnitine preparations were liquids. Then DL-carnitine HCl became available in bulk quantities, and later pure L-carnitine.
Initially, carnitine deficient patients were supplemented with DL-carnitine HCl. But after publications that haemodialysed patients got problems after consumption of pure DL-carnitine [103], this compound was banned, and L-carnitine became the preferred drug.
There are several methods for the industrial production of L-carnitine. Chiral separation of DL carnitine, direct organic synthesis [104] or the conversion of crotonobetaine, deoxycarnitine or dehydrocarnitine by bacterial preparations. The latter methods are most widely used [105].
This has been written as a tribute to Professor Willem C H�lsmann (1928-2000) who was the first Director of our Department of Biochemistry of the Medical Faculty Rotterdam, later Erasmus University of Rotterdam from 1968 to 1988. The author Hans R. Scholte retires the end of the month September 2003 as professor of Biochemistry at Erasmus MC-University Medical Center Rotterdam, and thanks dr Ben Tilly and Ton Verkerk for their valuable indispensable help to get this work in cyberspace.
[ea= and colleague(s)]
1-Bremer J. Physiol Revs 1983;63:1420-79
2-Scholte HR ea in Gitzelmann R ea, eds, Carnitin in der Medizin, Schattauer, Stuttgart, 1987:21-59
3-Scholte HR ea in Seim H ea, eds Carnitine-Pathochemical Basics and Clinical Applications, Ponte Press, Bochum,1996:11-31
4-Bremer J in DeSimone C ea eds. Carnitine Today, Springer, New York,1997:1-37
5-McKusick ea. Online Mendelian Inheritance in Man. http://www3.ncbi.nlm.nih.gov/Omim/
6-Vaz FM, personal commun
7-Price N ea Genomics 2002;80:433-42
8-Murthy MSR ea.Proc Natl Acad Sci US1987; 84:378-382
9-Fraser F ea. Biochem J 1997; 323:711-8
10-Van der Leij FR ea. Biochem J 1999;341:777-84
11-Sekoguchi E ea. J Biol Chem (July 25, 2003)
12-Roe DS ea. Mol Genet Metab 2000;69:69-75
13-Ferdinandusse S ea.BBRC 1999; 263:213-8
14-Tamai I ea J Biol Chem 1998;273:20378-82
15-Tamai I ea. J Biol Chem 2000;275:40064-72
16-Wagner CA ea. Am J Physiol Renal Physiol 2000;279,F584-91
17-Stieger B ea. Biochem J 1995;309:643-7
18-Rodrigues Pereira R ea. Eur J Pediatr 1988;148:193-7
19-Xuan W ea. BBRC 2003;306:121-8
20-Enomoto A ea. J Biol Chem 2002; 277:36262-71
21-Rodriguez CM. Biol Reprod 2002;67:314-9
22-Lahjouji K ea. BBA 2002:1558:82-93
23-Paul HS ea. Eur J Biochem 1992; 203:599-605
24-Rebouche CJ ea. BBA 1980; 630:22-29
25-Vaz FM ea. Biochem J 2002;361:417-29
26-Gilde AJ. Circ Res. 2003;92:518-24
27-De Jong JW ea. BBA 1970; 197: 127-135
28-Pande SV ea. BBA 1970; 202: 43-48
29-Scholte HR ea, BBA 1975; 409:283-69
30-Krisans SK ea. J Biol Chem 1980 ;255:9599-607
31-Kee HJ ea. BBRC 2003 ;305:925-33
32-Ves-Losada A ea. Mol Cell Biochem 1996;159:1-6
33-Coleman RA ea. J Nutr.2002;132:2123-6
34-Wanders RJA ea. J Inher Metab Dis 1999;22:442-87
35-Scriver CR ea, eds, The Metabolic & Molecular Bases of Inherited Disease, 8th Ed, Vol. II,
McGraw-Hill, New York, 2001
36-Groot PHE ea. BBA 1973;316:124-35
37-Veerkamp JH ea. Biochem Med 1980;24:118-29
38-Cifone MG ea.Proc Assoc Am Physicians. 1997;109:146-53
39-Pastorino JG ea, J Biol Chem1993 ;268:13791-8
40-Mutomba MC ea. FEBS Lett. 2000 ;478:19-25
41-Virmani MA ea. Pharmacol Res 1995;32:383-9
42-Prickaerts J ea Neurochem Int 1998;33:435-43
43-Mazzio E ea.Biochem Pharmacol 2003;66:297-306
44-Scallet AC ea. Ann N Y Acad Sci 2003 ;993:305-12
45-Virmani A ea. Ann N Y Acad Sci 2003;993:197-207
46-Gadaleta MN ea. Eur J Biochem 1990;187:501-6
47-Aureli T ea. Brain Res 1994;643:92-9
48-Castorina M, Ferraris L. Life Sci 1994; 54:1205-14
49-Gadaleta MN ea.Biochimie 1998;80:863-70
50-Aureli T ea. Neurochem Res. 2000 ;25:395-9
51-H�lsmann WC ea. BBA 1985 ;847:62-6
52-H�lsmann WC ea. Cardioscience 1994 ;5:67-72
53-H�lsmann WC ea. Mol Cell Biochem 1996;156:87-91
54-H�lsmann WC. In De Simone C ea, eds. Carnitine Today, Springer, New York, 1997:163-9
55-Arduini A ea. J Biol Chem. 1992;267:12673-81
56-Arduini A ea. J Neurochem 1994;62:1530-1538
57-Arduini A ea. Transfusion. 1997;37:166-74
58-De Simone ea.Acta Vitaminol Enzymol. 1982;4:135-40
59-Jirillo E ea. Immunopharmacol Immunotoxicol 1991;13:135-46
60-De Simone C. Immunopharmacol Immunotoxicol. 1993 ;15:1-12
61-Famularo G ea. Immunol Today 1994;15:495-6
62-Winter BK ea. Br J Cancer 1995;72:1173-9
63-Arenas J ea. Early Hum Dev. 1998;53 Suppl:S43-50
64-Lohninger A ea.J Perinat Med 1996;24:591-9
65-Jeulin C ea. Hum Reprod Update 1996;2:87-102
66-Palmero S ea. Horm Metab Res 2000;32:87-90
67-Resh MD BBA 1999;1451:1-16
68-Dubelaar ML ea. J Appl Physiol. 1994 ;76:1636-42
69-Scholte HR ea J Clin Chem Clin Biochem.1990;28:351-7
70-Tein I ea. Pediatr Res 1990;28:247-255
71-Famularo G ea in De Simone C ea.Carnitine Today, Springer, New York; 1997:119-162
72-Calvani M ea. in Desnuelle C ea, eds. Mitochondrial Disorders, Springer, Paris, 2002:107-130
73-Ganapathy ME ea. J Biol Chem 2000 ;275:1699-707
74-Tein I ea Pediatr Res 1993;34:281-7
75-Marzo A ea. Gynecol Endocrinol 1994;8:115-20
76-B�hles H ea. Burns 1996 ;22:166
77-Heuberger W ea. Eur J Clin Pharmacol 1998;54:503-8
78-Farkas V ea. Biochem Pharmacol 1996;52:1429-33
79-Parvin R ea. Anal Biochem 1977; 79:190-201
80-Takahashi M ea. Clin Chem1994;40:817-21
81-Roe CR. Prog Clin Biol Res 1990;321:383-402
82-Vreken P ea. Adv Exp Med Biol 1999;466:327-37
83-Carpenter KH ea. Clin Chim Acta. 2002;322:1-10
84-DiMauro in Desnuelle G ea, eds.Mitochondrial Disorders, Springer, Paris, 2002:307-319
85-De Jong JW ea, eds. The Carnitine System. Kluwer, Dordrecht, 1995
86-Derosa G ea. Clin Ther 2003;25:1429-39
87-Plioplys AV ea. Neuropsychobiology 1997;35:16-23
88-Graziano F ea. Brit J Cancer 2002;86:1854-7
89-Neri S ea. Neuropsychobiology 2003;47:94-7
90-Van Oudheusden LJ ea. Prostaglandins Leukot Essent Fatty Acids 2002;67:33-8
91-Torriolli MG ea. Am J Med Gen 1999;87:366-8
92-Montgomery SA ea. Int Clin Psychopharmacol 2003;18:61-71
93-Sorbi S ea. Clin Neuropharmacol 2000;23:114-8
94-Vermeulen RCW ea. Psychosom Med, 2003 in press
95-Rehman HU. Postgrad Med J 1999;75:451-2
96-Van Oudheusden LJ, personal commun
97-Vermeulen RCW, personal commun
98-Hoffmann GF, Neuropediatrics 1996;27:115-123
99-Walter JH. J Inher Metab Dis 2003; 26:181-188
100-Bellinghieri G ea. Am J Kidney Dis 2003 ;41 Suppl 1:S116-22
101-Steinman TI ea. Nephrol News Issues. 2003;17:28-30
102-Miller B ea.Am J Kidney Dis. 2003;41 Suppl:S44-8
103-De Grandis D ea.J Neurol Sci 1980;46:365-71
104-Jain RP ea.Tetrahydron Lett 2001;42:4437-40
105-Europe’s network of patent worldwide databases. http://ep.espacenet.com/
Full references and abstracts see: http://www.ncbi.nlm.gov/PubMed
![]() Back to Molecule of the Month page. [DOI:10.6084/m9.figshare.5437231]
Back to Molecule of the Month page. [DOI:10.6084/m9.figshare.5437231]