
This chapter is based on several publications with several authors and especially Jaenicke. L.; Marner. F.-J..
In contrast to the ionones that are apparently widespread in nature, the irones so far have been found only in orris root oil and more recently in oak moss extracts [7]. Looking at the structural characteristcs of irones as compared to ionones their rare occurence is not very surprising. The ionones have a normal terpenoid skeleton and can be thought of as oxidative cleavage products of caratenoids, which indeed often are sources of these compounds. The structure of irones, in contrast, does not fellow the isoprene rule. In particular, the methyl group at C(2) of the ring represents a feature which requires an explanation. Nevertheless the terpenoid nature of the irones is beyong doubt. Nobody ever asked how the irones originate in the plant but for one attempt to find ring-methylated caratenoids in rhizome extracts which, however, failed [8].
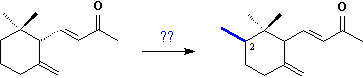
Certainly the C14-ketones
are not the original secondary matabolites of the
plants. The producers of orris oil have long known that freshly
harvested rhizomes do not contain any irones
at all and that the scent develops gradually reaching its maximum after
3 to 4 years of storage of the peeled root-stocks [9];
yet the reason for this development - whether enzymatic
or by other means - has remained obscure.
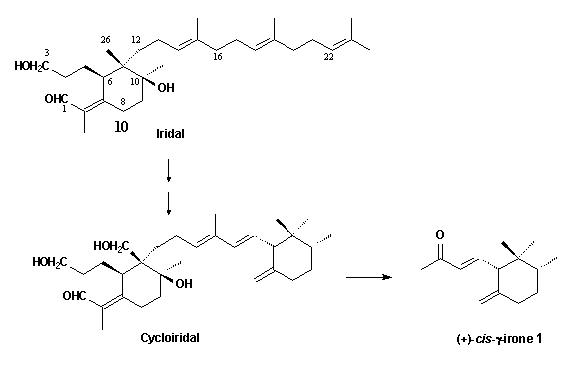
These C14-ketones
are derived from oxidative degradation of methylated bicyclic triterpenoids,
the cycloiridals, which are found together with the
monocyclic iridals 10 in roots and rhizomes of several Iris
species.
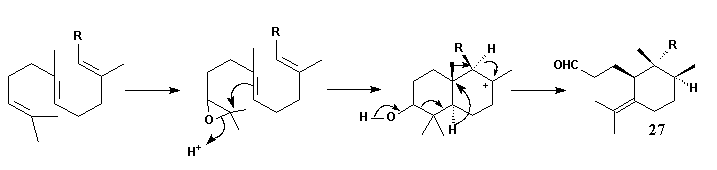
Epoxidation of squalene and subsequent cyclisation leads to a bicyclic ionic intermediate which rearranges in the way shown to form the skeleton 27 of the Iris triterpenoids. By this scheme not only the unusual substitution pattern but also the stereochemistry of the compounds may be explained. C(1) and C(10) have to be oxidized - the latter with inversion of configuration - and C(3) must be reduced to obtain the basic compound iridal 10.A more probable pathway for formation of Iridal 10:
Mechanism of Irone Ring Formation:
Finally the question posed at the beginning has to be answered: when and how the extra methyl group introduced into the unsatured C(22) position of the iridals? The irone moiety, biosynthetically, is formed by transfer of a Me group, probably via S-adenosylmethionine (SAM), to the terminal double bond of an open-chain iridal and cyclization as shown by incorporation experiments with radioactively labelled methionine [10].
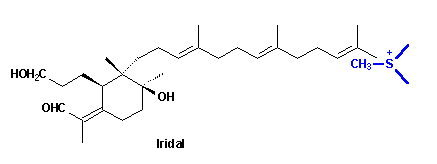
The most plausible mechanism for the addition would be one in which a cyclopropane cation is formed. The cyclization reaction may start immediately; alternatively, the cyclopropane cation may open to leave the positive charge either at C(22) or C(23). The former will give 22-methylene open chain compounds as end products. The latter, on the other hand, will lead to the cyclic compounds. The stereochemistry at C(22) of the cyclic products probably depends solely on the side from which SAM attacks the molecule. Attack from above leads to the 22 R-molecule; attack from bellow to the 22 S-isomer. Depending on the direction of the ring formation the 18 S- or 19 R-products will be formed.
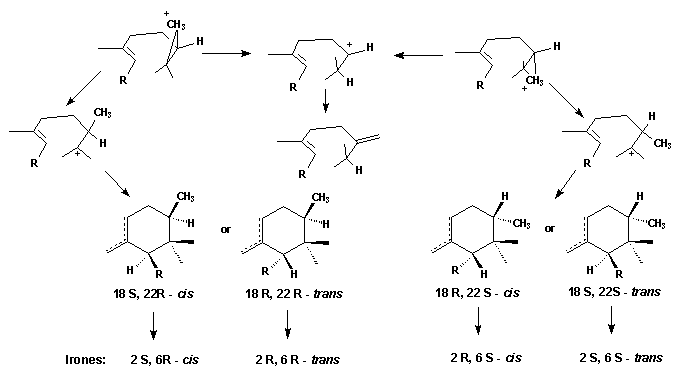
Thus the formation of all possible cis-
or trans-irone precursors is explained.
The simultaneous occurrence of both
2 R, 6 S-cis and 2 S, 6
S-trans-irones
within the same iris-oil implies, that the formation of the cyclopropane
cation is not stereospecific
- though one product seems to be preferred. The direction of ring closure,
however, and the twisting of the side chain R is unidirectional.
This may depend on the enzymatic 'environment'
during the cyclization e.g. the position
of a stabilizing negative charge or/and steric
effects.
![]()