The Co-C bond at the centre of all of the Vitamin B12 coenzymes has some very interesting properties, in particular it is a relatively weak bond which can potentially be broken in three ways during enzyme catalysis:
- Homolytically to generate a radical and a Co(II)
- Heterolytically to form a Co(I) and a carbocation
- Heterolytically to form a Co(III) and a carbanion
Mode 3, forming a Co(III) and a carbocation is observed during electrophilic attack on the Co-C bond, for example by Hg(II) or other electrophiles on methylcobalamin.
Thus it is the unique ability of the cobalt atom in the cobalamins (cobalt corrin complexes) to shuttle between three oxidation states, Co(I), Co(II) and Co(III) which is the enabling factor for all of the activities of Vitamin B12. A central issue is just how two different mechaisms have evolved for these various enzymes systems.
To quote Jack Halpern, a pioneer in our understanding of the strength of the Co-C bond: "The accessability of the CoIII, CoII and CoI forms of B12 confers great versatility on CH3 - B12 as a methylating agent, i.e. as a source of CH3-, CH3. and CH3+. Thus CH3 - B12 goes considerably beyond a "Grignard analogue" as a methylating agent."
The reaction whereby a hydrogen and a group on an adjacent
carbon atom exchange places 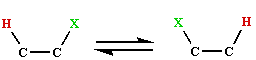 is considered to take place by way of a radical
mechanism, such rearrangements are very rare in organic
chemistry, indeed the only known instance is the Kharash-Urey
reaction. It is especially surprising that the reactions
involve specific configuration changes of the substrates. The
evidence for a radical mechanism comes principally from
observations of EMR (Electron Magnetic Resonance) spectra for several of the active enzyme
systems. Thus if the dioldehydrase (holoenzyme) is treated with
the substrate analogue 1,2-propoanediol, a radical signal
believed to be due to the 5'-deoxyadenosyl radical, together
with a low spin Co(II) signal is formed within milliseconds,
and persists whilst there is substrate present {T.H.Finley, J.Valinsky, K.Sato, and R.H.Abeles, 1972}. Abeles et al were the first researchers to propose the radical mechanism. Similar observations have been made for dioldehydrase-substrate, glycerol-dehydrase, ethanol-ammonia lyase, and the ribonucleotide reductase system.
is considered to take place by way of a radical
mechanism, such rearrangements are very rare in organic
chemistry, indeed the only known instance is the Kharash-Urey
reaction. It is especially surprising that the reactions
involve specific configuration changes of the substrates. The
evidence for a radical mechanism comes principally from
observations of EMR (Electron Magnetic Resonance) spectra for several of the active enzyme
systems. Thus if the dioldehydrase (holoenzyme) is treated with
the substrate analogue 1,2-propoanediol, a radical signal
believed to be due to the 5'-deoxyadenosyl radical, together
with a low spin Co(II) signal is formed within milliseconds,
and persists whilst there is substrate present {T.H.Finley, J.Valinsky, K.Sato, and R.H.Abeles, 1972}. Abeles et al were the first researchers to propose the radical mechanism. Similar observations have been made for dioldehydrase-substrate, glycerol-dehydrase, ethanol-ammonia lyase, and the ribonucleotide reductase system.
Since it is necessary to rigorously exclude dioxygen from any reaction involving free radicals, a plausible reaction mechanism {described in da Silva and Williams seminal text "The Biological Chemistry of the Elements"} is the following:
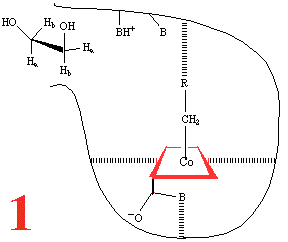
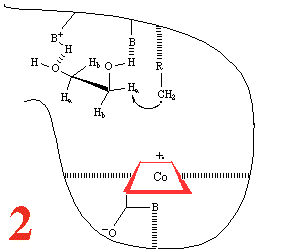 Step 1, and the substrate,
1,2-dihydroxyethane, approaches the holoenzyme, and is about to
plug the reaction 'bottle'. Then (Step 2) the
substrate binds and the cobalt-carbon bond breaks homolytically
to give the 5'-adenosyl radical and a Co(II) corrin, anchored
to the large coenzyme (are the amide groups involved?) It has
been suggested that the cobalt-carbon bond, already a rather
weak bond and readily subject to photolysis if the holoenzyme
is subjected to visible light, is further weakened by binding
of the metalloenzyme to the large coenzyme.
Step 1, and the substrate,
1,2-dihydroxyethane, approaches the holoenzyme, and is about to
plug the reaction 'bottle'. Then (Step 2) the
substrate binds and the cobalt-carbon bond breaks homolytically
to give the 5'-adenosyl radical and a Co(II) corrin, anchored
to the large coenzyme (are the amide groups involved?) It has
been suggested that the cobalt-carbon bond, already a rather
weak bond and readily subject to photolysis if the holoenzyme
is subjected to visible light, is further weakened by binding
of the metalloenzyme to the large coenzyme.
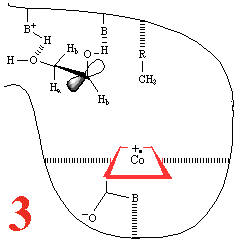
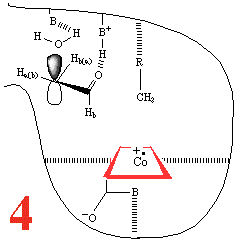 In Step 3,a hydrogen atom is
abstracted from the substrate by the 5'-deoxyadenosyl radical,
converting the dangling CH2 into a CH3
Then, Step 4, the enzyme base (B) and the acid
(BH+) cause assisted beta-hydroxy fragmentation,
that is loss of a water molecule.
In Step 3,a hydrogen atom is
abstracted from the substrate by the 5'-deoxyadenosyl radical,
converting the dangling CH2 into a CH3
Then, Step 4, the enzyme base (B) and the acid
(BH+) cause assisted beta-hydroxy fragmentation,
that is loss of a water molecule.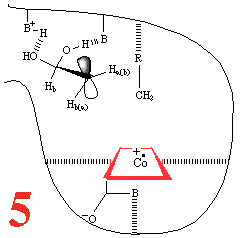
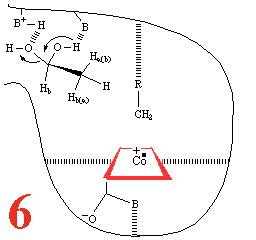 With Step 5, comes the
re-addition of a water molecule, resulting in a substrate
reorientation of C2 towards the 5'deoxyadenosyl
methyl. There must be competing C1 and C2
racemisation. A hydrogen atom abstraction occurs, with
competing C2 racemization. The cycle is completed
(Step 6), by substrate dehydration and release
of ethanal and a water molecule, so that the system can pick up
another substrate molecule.
With Step 5, comes the
re-addition of a water molecule, resulting in a substrate
reorientation of C2 towards the 5'deoxyadenosyl
methyl. There must be competing C1 and C2
racemisation. A hydrogen atom abstraction occurs, with
competing C2 racemization. The cycle is completed
(Step 6), by substrate dehydration and release
of ethanal and a water molecule, so that the system can pick up
another substrate molecule.
The enzymes that bind adenosyl cobablamin, and which catalyses group migrations (mutases) are all believed to be initiated by homolytic cleavage of the Co-C bond, forming an adenosyl radical and with the cobalmin in a Co(II) oxidation state.
There has been a flurry of interest in this subject now that details of the enzyme/coenzyme binding are starting to emerge from mutagenic and X-ray crystal studies of the enzyme.
See Ei-Ichiro Ochiai, for an early discussion of aspects of the radical mechanism. For a more recent discussion of this mechanism, and a description of how cobaloxime models assisted in the understanding, see Lippard and Berg.
For the methyl transfer reactions involving CH3-B12, it is likely that Co(I) is involved. The reaction catalysed by methionine synthase involves two methyl group transfers:- Me-cob(III)alamin + homocysteine ---> cob(I)alamin + methionine
- cob(I)alamin + methyltetrahydrofolate ---> Me-cob(III)alamin + tetrahydrofolate
Recent Molecular Mechanics Interest
Molecular mechanics and molecular dynamics simulations of porphyrins, metalloporphyrins, heme proteins and cobalt corrinoids.
H.M.Marques and K.L. Brown, Coord.Chem. Rev., 2002, 225, 123.
"The attention is focused on the use of molecular mechanics (force field) and molecular dynamics methods for the modeling of the structure of porphyrins (non-planar distortion, metal complexes and ligand interaction, spin states, N-substituent porphyrins, crystal structure effects, "designer" porphyrins), metalloporphyrins and hemoproteins (microperoxidases, Hb, myoglobin, peroxidases, cytochromes), and the cobalt corrinoids (derivs. of vitamin B12)."