![]()
WHY WATER? You're right if you think it's terribly difficult to select an aspect or two to discuss when the larger topic is water. There's all sorts of fascinating stuff! Everything from the role of the waters of hydration in antibody-binding to the effect of solvation for stabilizing protein structure to the dependence of water's dielectric and pH on temperature and pressure. This last topic is particularly exciting as we learn more about the thermophiles living at deep sea vents. And to paraphrase a guest I heard on a radio science-talk show recently, once we better understand the life living in our oceans, we'll be much better prepared to understand possible life on other planets.
But most importantly, because the aqueous environment influences all of the chemistry that takes place in it, when you are discussing water you get to address just about anything, while still ostensibly focusing on the substance itself.
Before we get to water at extremes of temperature and pressure, let's touch briefly on the oh so neglected role of water in our everyday life. And just to be sure we start out on a provocative note, let me here quote Felix Franks who told me
We immediately see the evident truth in this statement when we consider the complex thermodynamics of protein and peptide structural stability and the prominent role of solvent in influencing this equilibrium. Proteins unfold from their native fold upon the addition of urea or other denaturants, peptide helicity increases upon addition of TFE, and alcohol is even known to induce helical structure in regions that otherwise adopt beta-sheet conformations. While the helix inducing effects of alcohol upon peptide conformation have long been used to probe and enhance latent structure, thereby making it measurable by CD or NMR, a recent paper illustrates how this dependence of structure on solvent conditions may be harnessed to make a sort of 'conformational switch'. (See the reference from Kuntz). Successful molecular design or peptide engineering applications will necessarily include an assessment of solvation effects for conformational stability.
Turning from a macroscopic picture of the role of water and aqueous solutions in establishing the equilibria of proteinaceous substances to a more microscopic view, we can consider the surprising role played by discrete water molecules in antigen-antibody recognition. In a 1993 paper in Current Opinion in Immunology, Mariuzza and Poljak report a case in which interfacial water plays a critical role in determining the complementarity of the antigen and antibody surfaces. This work was illuminating, since previous work was leading to the conclusion that the packing density was lower at protein interfaces than in protein interiors. It also appeared that polar atoms at these interfaces were apparently tolerating the absence of neutralizing partners - far more than they do in protein interiors. In this work, a 1.8 angstrom crystal structure of an immune complex was studied and revealed that the water network was an integral part of the interface. As pointed out by these authors, the analysis of low resolution crystal structures may in fact lead to erroneous conclusions on the nature of the interface in these complexes. Furthermore, the evidence reported in this work points to the inescapable conclusion that the intermolecular complementarity essential for molecular recognition is not necessarily imparted solely by the large molecules.
And, contrary to what our notions of solvent entropy might lead us to expect, bound water may play a role in stabilizing the structure of single proteins as well as that of complexes. And, this bound water may, apparently, serve this function at the surface of the protein, and not solely while confined to the interior. (See Gronenborn.)
That's just the tip of the iceberg as far as far as water and its impact for biological systems at what is considered physiological temperature and pressure; the realm normally explored by biochemists and pharmacologists.
Much, however is being learned about the so-called "hot life", life found at thermal vents deep in the ocean floor. Reports have documented the existence of organisms at temperatures in excess of 110 deg. C (see Shock for a list of references). These organisms are referred to collectively as thermophiles. It is important to note the enormous temperature variation of sea water at these depths, with a documented range from 2 deg. C to 900 deg. C at depths of a few km where the high pressure precludes boiling. Hot life, or the existence of thermophiles, has been noted at temperature in excess of 100 deg. C at these elevated pressures, and it remains a matter of some dispute whether or not bacteria can exist at even higher temperatures still.
So, what are the properies of ambient sea water for organisms at deep sea thermal vents? We can expect these properties to influence the kinetics and thermodynamics of the biochemistry pertinent to this life, just as they influence the systems with which we are, perhaps, more familiar. We can begin to understand the fluid environment presented to thermophiles by considering the temperature dependence of a few properties of pure water.
A quick look at the following data from Shock show the dramatic effect of temperature on water's dielectric. Water at 25 deg. C and 1 atm pressure has the familiar dielectric near 80; however near water's critical point the dielectric has fallen to less than 10. For comparison, ethanol has a dielectric of roughly 25 at STP.
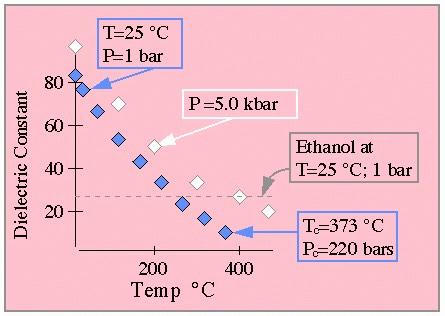 From Shock.
From Shock.
In this graph all white and blue diamonds pertain to water data points.
The blue diamonds are for pressures below the critical point where water
exists in two phases, while the white diamonds pertain to 5 kbar data. For
the low pressure data, two key data points are indicated. The grey dashed
line indicates the dielectric of ethanol at standard conditions. (This graph
is drawn to approximate that shown in the original paper which should be
consulted for quantitative work.)
The weak pressure dependence of this data should be noted. In order to understand the relevance of this sort of analysis to a complete understanding of ambient conditions at deep sea thermal vents, it should be understood that the pressure at these depths ranges from a few hundred bars to several kilobars. Shock should be consulted for additional details, data and a general groovy time.
Well, if all this is true, then water must have a pretty different structure at these T&P's than we are used to. Right? Not surprisingly, this is what is being found. Some of these results are shown in this graph:
.jpg) From Postorino
From Postorino
The pair distribution function for intermolecular OH atom pairs is shown
as a function of interatomic distance for water at 25 deg. C (red dashed
line) and 400 deg. C. (This graph is drawn to approximate that shown in
the original paper which should be consulted for quantitative work.)
This particular pair distribution function tells us the likelihood with which a H atom will be found at any distance from an O atom (The intramolecular data isn't shown here, but the water molecule does have the same structure at 400 deg. C as at 25 deg. C. This graph considers only intermolecular associations.) Immediately, then, we see that at room temperature it is relatively likely to find these atom pairs separated by 1.95 angstroms. This is the hydrogen bond. And then there are strong secondary structural phenomena, manifest as the damped oscillations that we see here out to beyond 5 angstroms, all due to the overwhelming hydrogen bonding behavior.
Looking at the high temperature data, it's clear that the hydrogen bonding peak, which dominates the room temperature data, is completely gone. The OO correlation function, not reproduced here, shows a slight broadening of the nearest-neighbor peak with temperature. The high temperature OO pair correlation function also yields a larger coordination number than is seen at room temperature. In other words, each water molecule is surrounded by more water molecules in its first solvation shell at 400 deg. C than at 25 deg. C. At 400 deg. C the packing is not restricted by the severe hydrogen-bonding geometrical constraints of the the room temperature fluid. This result is in line with the close packing behavior of most liquids. These results taken together with those for the dielectric phenomena described above begin to paint a picture of high temperature water. Water at high temperature exhibits many of the properties we associate with less polar liquids under standard conditions.
In order to understand something of the biochemistry that might pertain under extreme aqueous conditions, Shock has been estimating the stability of peptidic species at elevated temperatures and pressures. Specifically he has been asking questions like "How does the free energy of the peptide bond change with temperature and pressure?" A snapshot view of some of this work will serve to further the discussion of aqueous properties under extreme conditions while at the same time giving a hint for some of the very different biochemistry that will undoubtedly be revealed by further study in this area.
Considering only Glycine here, the free energy of interest here is that which determines the following equilibrium:
Glycine + Glycine = Glycylglycine + H2O
In other words, what is the free energy cost to form the peptide bond between two Glycine molecules? This equilibrium can be calculated from the free energies of formation for the individual species involved in this reaction, Glycine and Glycylglycine, and the temperature dependence of this free energy in the following way:
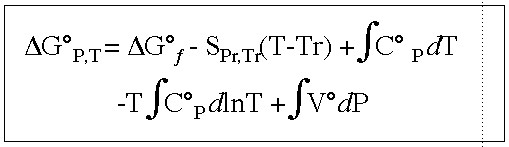
This equation has the usual host of characters. G represents free energy,
S is entropy, Cp is heat capacity, V is volume and P is pressure. If any
of this is murky please consult the original paper by Shock. What we're basically doing is getting the free energy (OK, so it's really
the standard partial molal Gibbs free energy) at any T&P from that at STP
by integrating over the temperature and pressure dependence of the heat
capacity and volume.
In order to do this, we need to know something about the way heat capacity and volume depend on the thermodynamic variables of T and P, respectively. We'll look at this first:
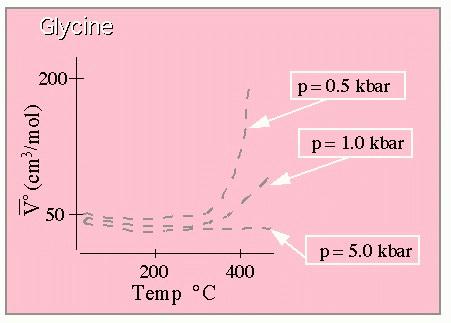
Standard partial molal volume for Glycine. From Shock. (This graph is drawn to approximate that shown in the original paper which
should be consulted for quantitative work.)
These results show the very large dependence of the molal volume on temperature at lower pressures, and the much weaker temperature dependence at extremely high pressures. Here are the results for the heat capacity:
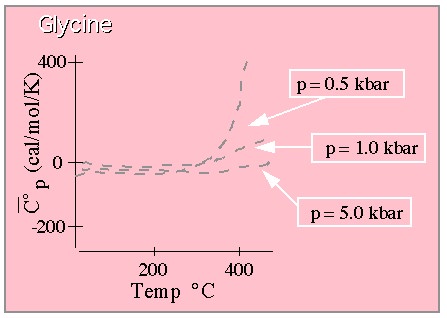
Standard partial molal heat capacity (constant pressure variety) for Glycine.
From Shock. (This graph is drawn to approximate that shown in the original paper which
should be consulted for quantitative work.)
Pretty much the same thing. A large T dependence is witnessed at lower temperature, but this behavior is convincingly squashed at high pressure.
The results for Glycine have been shown here. Analogous results for Glycylglycine are obtained from these by calculating a regression equation that relates, for instance, the partial molal volume of Glycine to that of Glycylglycine. (See Shock). Putting it all together with some other necessary data (like the free energy of formation at STP, and the entropy at STP; you got it, see Shock) the free energies of formation for both Glycine and Glycylglycine can be obtained at any temperature and pressure, and from these the free energy governing the equilibria may be directly obtained.
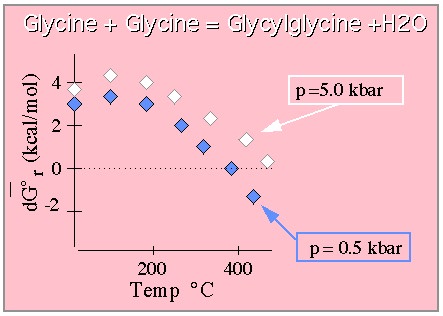
Free energy required to form the Glycylglycine dipeptide from the constituent
Glycine moieties. From Shock. (This graph is drawn to approximate that shown in the original paper which
should be consulted for quantitative work.)
The results indicate that while it costs free energy to form the dipeptide at lower temperatures and pressures (at STP the free energy of this reaction is roughly 3.5 kcal/mol), at extremes of temperature and pressure the dipeptide is the thermodynamically favored species. In any event, these results are consistent with those for many other dipeptides not covered here and predict that peptide bonds become more stable at elevated temperatures. This behavior just begins to hint at the interesting biochemical phenomena that will be revealed for life in water under extreme conditions where water has very different properties from those to which we are accustomed.
Afterword:
I would like to give appropriate acknowledgment to the various authors on whose work I have based this piece. In particular, this work is heavily motivated and influenced by the really interesting work of Everett Shock.
To read more about:
Water stabilizing biological structure:
Localization of Bound Water in the Solution Structure of the Immunoglobulin
Binding Domain of Streprococcal Protein G, G. Marius Clore and Angela M.
Gronenborn, J. Mol. Bio., 223, p. 853, 1992.
Conformational dependence on solution conditions:
Conformational Switching in Designed Peptides: the Helix/Sheet Transition
Roberta Cerpa, Fred Cohen, Irwin Kuntz, Folding & Design, 1( 2), p. 91, 1996.
Water at extremes:
Chemical Environments of Submarine Hydrothermal Systems, Everett Shock,
in Origins of LIfe and Evolution of the Biosphere, 22, 67-107, 1992.
Stability of Peptides in High-Temperature Aqueous Solutions, Everett Shock, Geochim et Cosmochim Acta, 56, p. 3481, 1992.
The Interatomic Structure of Water at Supercritical Temperatures, P. Postorino et al., Nature, 366, p. 668, 1993.
Write me: Shawn Huston Kenner (Oct 1996)
![]() Back to Molecule of the Month page. [DOI:10.6084/m9.figshare.5469604]
Back to Molecule of the Month page. [DOI:10.6084/m9.figshare.5469604]