Research
Most biochemical reactions depend on enzyme catalysis, and understanding how enzymes 'work' at the molecular level is a fundamental problem. Enzymes are remarkably efficient and specific catalysts, but despite intensive experimental investigations, the detailed origins of their rate accelerations remain unclear.
This question is of crucial importance in biology, and also for the development of protein catalysts for practical applications.
Better understanding is vital for analysing the activities of mutant or designed proteins, and for the design of inhibitors as pharmaceutical lead compounds.
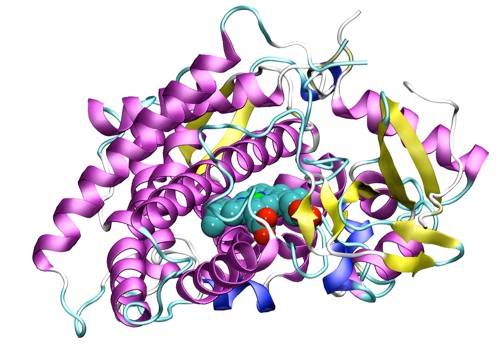
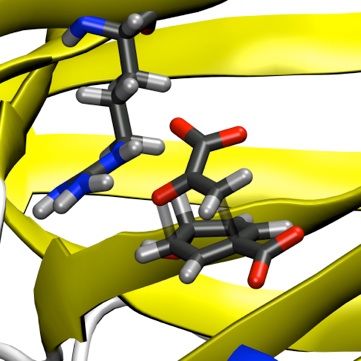
Computer simulations are a good way to study enzyme reactions. They can provide information which is often inaccessible experimentally, such as details of unstable species (for example transition states and reaction intermediates) and on energetic contributions to catalysis. Simulating an enzyme reaction is a challenging problem, and requires the use of specialized techniques. An effective approach is to use combined quantum mechanical/molecular mechanical (QM/MM) methods. Molecular mechanics methods can be used to study protein conformational changes, dynamics and binding, but generally they can't be applied to processes involving the breaking or making of chemical bonds. For chemical reactions a quantum mechanical description is needed, which can be achieved by the combined QM/MM approach. The small QM region contains the groups involved in the reaction (e.g. the catalytic residues and the substrate(s)) and is influenced by the surroundings (represented more simply by standard molecular mechanics), so including the effects of the environment. In this way the reaction in an enzyme can be studied, and contributions of individual groups can be analysed.