4.
Computer Simulation
4.1. Introduction
Computer simulations have been undertaken of the gas phase chemistry for all microwave and hot filament activated diamond CVD gas mixtures investigated by MBMS. Simulations involved the use of the CHEMKIN II suite of computer packages. The Chemkin interpreter was used to process reaction mechanistic data into a form recognisable to the SENKIN package. SENKIN is a zero dimensional kinetics simulation package used to predict species mole fractions, for a given gas mixture under a given set of conditions.
Initial studies focused on CH4/CO2 gas mixtures, covering the entire range of mixing ratios (i.e. 100%CO2-100%CH4). However, these investigations have also been extended to include 36 additional H/C/O containing gas mixtures, allowing prediction of the diamond domain boundaries of the Bachmann C-H-O atomic phase diagram (see Section 1.14.1).
A reaction mechanism for the coupling of C and S has been proposed that has enabled simulations to be carried out for xH2S/1%CH4/H2 and xH2S/51%CH4/49%CO2 (x = 0-1%). In addition, species mole fractions have been predicted for 0.5%H2S/1%CH4/H2 and 1%CS2/H2 gas mixtures, as a function of gas temperature, Tgas. Such studies of sulfur containing gas mixtures have led to the proposal of a parameter, Fs for the prediction of S-incorporation into diamond films. Fs has been defined as the product of SENKIN calculated mole fractions of CH3 and CS, [CH3]´[CS].
4.2 Overview of CHEMKIN II
CHEMKIN II is a FORTRAN chemical kinetics package for the analysis of gas-phase chemical kinetics. The package can be used to model gas phase reactions such as those occurring in combustion or diamond chemical vapour deposition.
The package is made up of two major components: an Interpreter and a Gas-Phase Subroutine Library. The Interpreter takes a user‑selected reaction mechanism and converts it into a form that the modular subroutines contained in the Gas-Phase Subroutine Library can understand. These subroutines are used by application codes that perform calculations using the output from the Interpreter, an input file (providing instructions on the calculations to be carried out) and in many cases, data contained within a reference database.
The application codes are used to perform calculations relevant to specific chemical problems (e.g. different reactors). The Interpreter and the SENKIN application will be more fully explained in the following sections. Note that text in Arial font refers to directory or file names, whereas typed commands are presented in Italic text.
4.3. Files
The actual Chemkin Interpreter and SENKIN program files were obtained from Sandia National Laboratories (California, USA), with permission, via the Internet.
4.3.1. Setting up a directory for files
CHEMKIN input, output and database files were located in a directory on a UNIX computer system named grendel [[1]]. Grendel is a Beowulf system; it consists of 16 computers, each of which has two 400 MHz Pentium II processors, 512 Mbytes of memory and 10 Gbytes of disk and runs the Redhat distribution of Linux. A complete guide to using UNIX can be found within Reference [2], however essential commands needed to run CHEMKIN will be given here.
When a user logs onto grendel (using SSH Secure Shell Client) for the first time they will be looking at the contents of the directory called username (e.g. jp5147). In order to create the directory chemkin the user types mkdir chemkin. The executable application programs (e.g. chem.exe and senk.exe) are located on grendel in a separate directory (/usr/local/bin), which is linked to the directory chemkin automatically.
4.3.2. File Handling
Short input files (e.g. for applications such as SENKIN) were created using the Pico editor. For example the file senk.inp was created by typing pico senk.inp. If a file called senk.inp exists, this command would open it (thus allowing the examination of the contents of files such as output files). Longer input files (e.g. chem.inp used by the Chemkin Interpreter) were edited using the Word package (on a PC) before being saved as a text file (e.g. chem.txt). This text file can then be renamed chem.inp and transferred onto grendel using a FTP package (SSH Secure File Transfer Client). The file can also be transferred back to a PC for editing.
4.4. CHEMKIN Interpreter (chem.exe)
The Interpreter took the reaction mechanism supplied by the user and extracted the thermodynamic data for each species from the thermodynamic database. These data were used to calculate reverse rate constants for all the reversible reactions included in the input reaction mechanism (see Section 4.6). The Interpreter then generated two output files. The first was human-readable, and listed the reactions along with any errors (if any) associated with each. The second was a binary link file that could be understood by application codes such as SENKIN. A diagram illustrating the structure of the CHEMKIN package and its relationship to the Gas-Phase Subroutine Library is shown below. File names of each unit (as saved on grendel) are given in brackets.
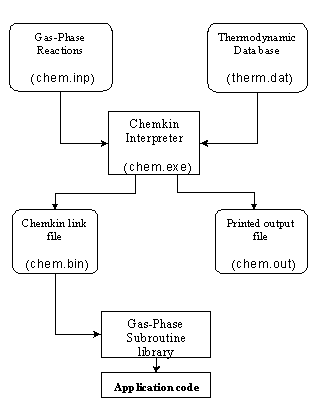
Figure 4.1. Schematic diagram showing the structure
of the Chemkin package and its relationship to an application code (adapted
from Reference [3]).
4.5. Input Gas-Phase Reactions
(chem.inp)
This section describes the structure and contents of the CHEMKIN reaction mechanism input file, chem.inp. Firstly, an example input file will be presented and explained, before the actual mechanisms used for gas phase chemical kinetics simulations are discussed.
Below is a breakdown of an example chem.inp file. First all the elements, which appear in the mechanism, are specified:
ELEMENTS
H C
END
then all the species:
SPECIES
H2 H C
CH CH2 CH3
CH4
C2H C2H2 C2H3
C2H4 C2H5 C2H6
END
When the code is run, chem.exe extracts the thermodynamic data for these elements and species from the thermodynamic data base. The reactions are then listed in the form:
REACTIONS
H+CH<=>C+H2 1.100E+14
.000 .00
H+CH2(+M)<=>CH3(+M) 2.500E+16
-.800 .00
LOW /
3.200E+27 -3.140 1230.00/
TROE/ .6800
78.00 1995.00 5590.00 /
H2/2.00/ CH4/2.00/ C2H6/3.00/
END
In this example
both of the reactions are reversible as signified by the symbol <=>. The three columns of numbers contain the
Arrhenius parameters for the reactions. The first reaction is shown below as
Reaction 4.1.
kF
H +
CH ![]() C + H2 Reaction 4.1
C + H2 Reaction 4.1
kR
In this case the forward rate
constant, kF, is calculated from the Arrhenius parameters
thus,
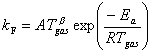 Equation
4.1
Equation
4.1
where A is the
pre-exponential factor (1.1´1014) and b and Ea are the
temperature exponent and activation energy (both zero in the case of this
particular reaction), respectively. It
should be noted that the default units for the Arrhenius parameters A
and E are [cm mole sec K] and [cal/mole] respectively.
The second
reaction is a pressure dependent fall-off reaction. The first set of Arrhenius
parameters (A¥, b¥
and E¥) provided is for the high-pressure
limit. At this limit the appropriate
description of the reaction is H + CH2 ¾ CH3. The second set (A0, b0 and E0) refers
to the low-pressure limit i.e.
LOW / 3.200E+27
-3.140 1230.00/
The appropriate description for the reaction at the low-pressure limit is H + CH2 + M ¾ CH3 + M. The two sets of Arrhenius parameters give two rate constants, one (k0) at the low-pressure limit and one (k¥) at the high‑pressure limit. The reaction at any intermediate pressure (i.e. within the “fall-off” region) is described as H + CH2 (+ M) ¾ CH3 (+ M), and has a rate given by
k
= k¥ (Pr /1+Pr)F Equation
4.2
Where Pr is the reduced pressure given by Equation 4.3, in which [M] is the concentration of the mixture and Equation 4.4 defines F.
![]() Equation
4.3
Equation
4.3
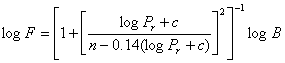 Equation
4.4
Equation
4.4
where c and n are constants dependent on B and
![]() Equation 4.5
Equation 4.5
In the input file these parameters (a, B3, B1 and B2) are defined in the form
TROE/ .6800 78.00
1995.00 5590.00 /
If a Lindemann fall-off reaction is used, only A0, b0 and E0 are required therefore this line is omitted.
The next line in the input file gives the enhanced third body efficiencies of the species included in the reaction mechanism that could act as third bodies in the reaction (i.e. +M), in the form
H2/2.00/ CH4/2.00/ C2H6/3.00/
The end of the reaction mechanism is signified by the key word END. When a line in the input file starts with ! the program ignores the rest of the line. Comments can therefore be inserted into the program, for example for the purpose of reaction categorisation.
The reaction mechanisms used for the present work are presented as a combined chem.inp file in Appendix 4. The sources of the reaction data will be discussed in the following sections.
4.5.1. Gri-Mech 3.0 Reaction Mechanism
All of the C, H and/or O containing species reactions and temperature dependent rate constants used in the SENKIN calculations presented in Chapter 5 were obtained from the GRI-Mech 3.0 reaction mechanism [[4]]. GRI-Mech 3.0 was developed by the Gas Technology Institute (Illinois, USA) to be an optimised mechanism for the modelling of natural gas combustion. The mechanism was obtained from Reference 4 via the Internet. All the thermodynamic species properties required to run this mechanism were obtained from the Chemkin Thermodynamic database, as described in Section 4.6.
4.5.2. C-S linking Reaction Mechanism
A reaction scheme was constructed to allow the gas phase coupling of C and S [[5]]. This was intended to allow the modelling of S-containing gas mixtures (Chapters 6 and 7). A stepwise mechanism was conceived based on the overall Reaction 4.2.
kF
CH4
+ 2H2S ![]() CS2 + 4H2 Reaction 4.2
CS2 + 4H2 Reaction 4.2
kR
The sulfur species reactions and their temperature dependent rate constants are presented in Table 4.1. In this scheme, reaction is initiated by breakdown of H2S, both thermally and also via hydrogen abstraction, to form SH (Reactions 1 and 2). CH3 (produced from CH4 via analogous mechanisms) combines with SH to form either CH3SH or CH2S (Reactions 3 and 4), which then undergo successive H-abstraction reactions (Reactions 5-9) to form CS and finally CS2 (via Reaction 10). Additional reactions form S (from SH, Reactions 11-13) and S2 (Reactions 14-16). The kinetic data presented are in the form of Arrhenius equation parameters which allow calculation of the rate constant of the forward reaction, k1, (as each is written) via Equation 4.1. Information regarding reactions involving S containing species was obtained from the literature, with rate data for analogous O containing species reactions used when none for S were available. These reactions are labelled (*) in Table 4.2.
Number |
Reaction |
A/ (cm3 mol-1 s-1) |
b |
Ea /(cal mol-1) |
Reference |
|
1 |
H2S ¾ SH+H |
7.632´1014 |
0.00 |
82155.00 |
|
|
2 |
H2S+H ¾ SH+H2 |
1.078´1013 |
0.00 |
2969.67 |
6 |
|
3 |
SH+CH3 ¾ CH3SH |
9.998´1012 |
0.00 |
0.00 |
|
|
4 |
SH+CH3 ¾ H2+CH2S |
1.018´1012 |
0.00 |
0.00 |
[8] * |
|
5 |
H+CH3SH ¾ CH3+H2S |
6.926´1012 |
0.00 |
1664.00 |
|
|
6 |
H+CH3SH ¾ CH3S+H2 |
2.903´1013 |
0.00 |
2593.00 |
9 |
|
7 |
H+CH3S ¾ CH2S+H2 |
1.988´1013 |
0.00 |
0.00 |
[10] * |
|
8 |
H+CH2S ¾ HCS+H2 |
5.252´1012 |
1.77 |
2989.29 |
10 * |
|
9 |
H+HCS ¾ H2+CS |
1.211´1014 |
0.00 |
0.00 |
10 * |
|
10 |
SH+CS ¾ H+CS2 |
3.232´1010 |
1.50 |
495.00 |
[11] * |
|
11 |
H+SH ¾ H2+S |
1.301´1013 |
0.00 |
0.00 |
|
|
12 |
CH4+S ¾ CH3+SH |
2.042´1014 |
0.00 |
19796.00 |
|
|
13 |
H2S+S ¾ 2SH |
5.704´1014 |
0.00 |
15045.00 |
|
|
14 |
2S+M ¾ S2+M |
1.200´1017 |
-1.00 |
0.00 |
4* |
|
15 |
SH+S ¾ S2+H |
2.409´1013 |
0.00 |
0.00 |
|
|
16 |
2SH ¾ H2+S2 |
3.012´1010 |
0.00 |
0.00 |
15 |
Table 4.1. Reaction scheme
proposed for C-S coupling within sulfur containing diamond CVD gas mixtures.
The thermodynamic data required to run CHEMKIN were found to be unavailable for a number of the species included in this S-C coupling mechanism. It was therefore necessary to generate such data as described in Section 4.6.1.
4.5.3. Leeds’ Combined Combustion Reaction Mechanism
All additional S/O/H containing species (e.g. SO, SO2, HSO, etc…) reactions were obtained from the Leeds’ reversible methane/nitrogen/sulfur combustion mechanism obtained from Reference [16] via the Internet. This mechanism has been successfully used to model methane/oxygen/argon laminar flames doped with ammonia and sulfur. However, 70% of the S/O containing species reactions within this scheme are either estimates, or based on low temperature measurements [[17]]. The mechanism also included all the required relevant species thermodynamic properties (Section 4.6.1).
4.6. Database of Species Thermodynamic Properties (Therm.dat)
Species thermodynamic properties are required in order for the Chemkin Interpreter (Section 4.4) to calculate reverse rate constants, for the reactions contained in the input mechanism (Section 4.5). For a given reaction, the reverse rate constant, kR, is related to the forward rate constant, kF, through the equilibrium constant, Kc, as
![]() Equation
4.6
Equation
4.6
Although Kc is given in concentration units, the equilibrium constant is more readily determined from the thermodynamic properties in pressure units; they are related by
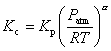 Equation
4.7
Equation
4.7
where Kp is the equilibrium constant expressed in pressure units and Patm refers to a pressure of 1 atm. The coefficient a corresponds to the sum of the product stoichiometric coefficients minus the sum of the reactant coefficients. The equilibrium constant is obtained from the change in standard state entropy, DS°, and enthalpy, DH°, that occurs in passing completely from reactants to products. Thus,
![]() Equation
4.8
Equation
4.8
The Chemkin thermodynamic database [[18]] contains data for species S°, H° and specific heats, Cp. The data are stored in the form of polynomial fits to Cpº/R, Hº/RT, and Sº/R as functions of temperature. There are seven coefficients for each of two temperature ranges (e.g. 298-1000 K and 1000-5000 K), which are defined in Equations 4.9-4.11. The high and low temperature ranges are linked by a common temperature (e.g. 1000 K).
![]() Equation
4.9
Equation
4.9
![]() Equation
4.10
Equation
4.10
![]() Equation 4.11
Equation 4.11
Each database entry contains the species name, its elemental composition, electronic charge and phase (i.e. gas, liquid or solid). The lower, upper and common temperatures are defined (in units of Kelvin) for the two temperature ranges. The coefficients are then listed, first for the upper (a1-a7) and then the lower (a1-a7) temperature ranges. The general layout is shown below, together with an example entry.
|
Species Name |
Date |
Composition |
Phase |
Low T |
High T |
Common T |
1 |
||
|
a1 |
a2 |
a3 |
a4 |
a5 |
2 |
||||
|
a6 |
a7 |
a1 |
a2 |
a3 |
3 |
||||
|
a4 |
a5 |
a6 |
a7 |
|
4 |
||||
C2H2
121386C 2H 2
G 0300.00 5000.00
1000.00 1
0.04436770E+02 0.05376039E-01-0.01912817E-04
0.03286379E-08-0.02156710E-12 2
0.02566766E+06-0.02800338E+02 0.02013562E+02
0.01519045E+00-0.01616319E-03 3
0.09078992E-07-0.01912746E-10 0.02612444E+06 0.08805378E+02 4
4.6.1. Thermodynamic Properties of S Containing Species
All H/C and O containing species thermodynamic properties required for use alongside the GRI-Mech 3.0 reaction mechanism were already present within the Chemkin thermodynamic database [18]. However, additional data required for use along side the C-S linking and Leeds’ combustion mechanisms (see sections 4.5.2 and 4.5.3) were found to be absent. Although the Leeds mechanism included related thermodynamic data, the C-S linking reaction mechanism included four species (CH3SH, CH3S, CH2S and HCS) for which the required data were not available. It was therefore decided to use a computational approach to estimate the required values.
The Gaussian 98 computational chemistry program [[19]] was used to generate minimised geometries for CH3SH, CH3S, CH2S and HCS, and these were then used to calculate the required enthalpy and Gibbs free energy corrections (with both calculations using the MP2/6-311G** basis set) for the range of temperatures of interest (298-6000 K). The data for each species were then scaled to the values at 298 K, using calculation of DGreac and DHreac for a number of isodesmic reactions (i.e. same number of bonds present on either side of the reaction) involving CH3SH, CH3S, CH2S and HCS. Figure 4.2 shows plots of enthalpy corrections which are (a) uncorrected and (b) scaled to value of H calculated at 298 K, H298, for HCS.
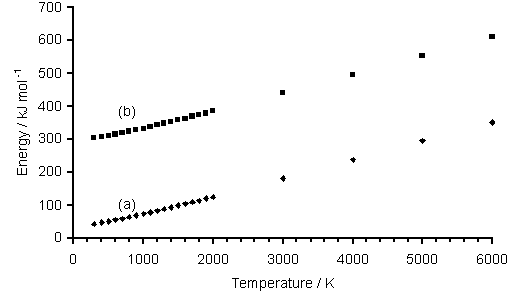
Figure 4.2. Plot of Gaussian calculated enthalpy
corrections vs temperature for (a) raw uncorrected data and (b) data
scaled to value calculated for H298.
An example follows for the calculation of H298 for HCS. Firstly, DHreac was obtained for the reaction of CS and H2 forming HCS and H, by summing the change between products and reactants of electronic energy and enthalpy energy correction (using CCSD(T)/cc‑pVDZ and MP2/6-311G** electronic structure calculations, respectively [[20]]). H° for HCS was then obtained via a Hess-type calculation using literature values [[21]] of H for CS, H2 and H. Similarly, G for HCS was calculated, leading to a value of S for the species being obtained. Similar calculations allowed the determination of H and S, at 298 K, for CH3SH, CH3S, CH2S and HCS (as presented in Table 2). Thus, data for standard heat capacity, enthalpy and entropy (as functions of temperature) were estimated for the four species, in the form of polynomial fits to Cpº/R, Hº/RT, and Sº/R as functions of temperature. A plot of Hº/RT vs temperature for HCS is presented in Figure 4.3. Calculated values of entropy and enthalpy at 298 K are given in Table 4.2, with H298 data being compared with literature data.

Figure 4.3. Plot of H°/ RT vs T. for
scaled HCS enthalpy corrections.
Temperature range / K |
Coefficient |
CH3SH |
CH3S |
CH2S |
HCS |
|
298-1000 |
a1 |
1.8873 |
1.5421 |
2.4953 |
3.5537 |
|
a2 |
1.5460×10-2 |
1.5180×10-2 |
6.1100×10-3 |
2.0700×10-3 |
|
|
a3 |
-5.6022×10-6 |
1.0000×10-5 |
4.3754×10-6 |
2.4041×10-6 |
|
|
a4 |
-1.8247×10-9 |
3.8581×10-9 |
-9.6164×10-9 |
-3.7711×10-9 |
|
|
a5 |
1.5779×10-12 |
-4.0429×10-13 |
4.2503×10-12 |
1.4823×10-12 |
|
|
a6 |
-3.9260×103 |
1.3532×10-4 |
1.1906×104 |
3.5321×104 |
|
|
a7 |
1.5501×101 |
1.6659×101 |
1.1629×101 |
7.4244 |
|
|
1000-5000 |
a1 |
4.5472 |
3.6110 |
3.4681 |
3.7778 |
|
a2 |
1.0180×10-2 |
8.9200×10-3 |
6.2500×10-3 |
2.8700×10-3 |
|
|
a3 |
-3.7614×10-6 |
-3.6176×10-6 |
-2.5499×10-6 |
-1.0604×10-6 |
|
|
a4 |
6.3502×10-10 |
6.8596×10-10 |
4.8585×10-10 |
1.7918×10-10 |
|
|
a5 |
-4.0058×10-14 |
-4.9459×10-14 |
-3.5164×10-14 |
-1.1311×10-14 |
|
|
a6 |
-4.8540×103 |
1.2974×104 |
1.1504×104 |
3.5163×104 |
|
|
a7 |
8.9853×10-1 |
6.4563 |
5.9022 |
5.7494 |
|
|
H298K / kJ mol-1 |
Calculated |
-22.69 |
121.21 |
107.60 |
303.34 |
|
Experiment |
-22.861) |
121.002) |
118.863) |
301.143) |
|
|
S298K / J mol-1 K-1 |
255.83 |
244.42 |
231.27 |
235.01 |
|
1)
Reference [22].
2)
Derived using H298K
for CH3SH from Reference 22 along with the experimentally determined bond dissociation enthalpy of CH3S-H
(Ref [23])
and H298K of H
(Ref 21)
3)
Reference [24].
Table 4.2. Polynomial fit coefficients for plots of Hº/RT,
and Sº/R as functions of temperature. The polynomial functions are described in Equations 4.10 and
4.11. Note that stated values of H298
and S298 can be converted from units of kJ mol-1
to kcal mol-1 by dividing by a factor of 4.1868.
4.7. Running the CHEMKIN Interpreter
After logging onto grendel (using SSH Secure Shell Client) the interpreter program is run by typing the word chem. The chem.exe code then reads chem.inp and therm.dat and two files are produced (chem.out and chem.bin). The chem.out file should then be checked for errors.
4.7.1. Checking the chem.out file
In order to check the chem.out file for errors type the command pico chem.out. This opens the file using the editor Pico. The page can then be scrolled down (ctrl+v) and up (ctrl+y) in order to find any error messages. If none are present the following statement will be at the end of the file:
NO ERRORS FOUND ON
INPUT...CHEMKIN LINKING FILE WRITTEN
The Chemkin linking file referred to is chem.bin, which is then used by the application codes such as SENKIN or PREMIX. Example chem.inp and chem.out files are given in section 6.
4.8. SENKIN (senk.exe)
SENKIN is a FORTRAN computer program which predicts the time-dependent chemical kinetics behaviour of a homogeneous gas mixture in a closed system. The program takes a starting set of conditions (pressure, temperature, species mole fractions) and then calculates how these change over time by integrating over small time steps. The program can deal with five different chemical problems:
· An adiabatic system with constant pressure (type A)
· an adiabatic system with constant volume (type B)
· an adiabatic system with the volume being a specified function of time (type C)
· a system where the pressure and temperature are constant (type D)
· a system where the pressure is constant and the temperature is a specified function of time (type E)
SENKIN takes the Chemkin linking file (chem.bin) and a keyword input file (senk.inp) and uses the Gas-Phase Subroutine Library to produce a binary output file (save.bin) and a readable output file (tign.out). A diagram showing how SENKIN and the CHEMKIN Interpreter are related is shown below.
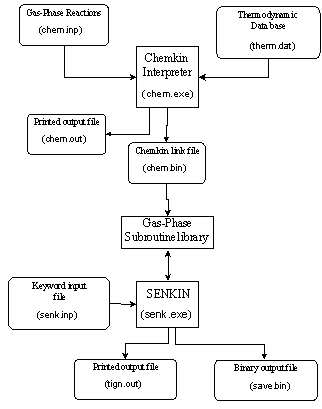
Figure 4.4. Relationship of
the SENKIN code to the Chemkin Interpreter and the associated input and output
files (adapted from Reference [25])
4.8.1. Keyword input file (senk.inp)
This file tells the program which of the five possible problems it is to solve, the initial conditions and the integration required (total reaction time and time step length). The problem type is specified by entering one of the following keywords into senk.inp.
CONP – The solution will be obtained with the pressure held constant at the initial value, as in type A.
CONV – The solution will be obtained with the volume held constant at the initial value, as in type B.
VTIM – The solution will be obtained with the volume as a function of time, as in type C. Subroutine VOLT (TIME, VOL, DVDT) must be provided to specify volume.
CONT – The solution will be obtained with both the pressure and volume held constant at the initial value, as in type D.
TTIM – The solution will be obtained with pressure held constant and the temperature a function of time. Subroutine TEMPT (TIME, TEMP) must be provided to specify temperature.
The initial conditions are specified using the following keywords.
TEMP – The initial temperature of the gas mixture in K (e.g. TEMP 2000)
PRES – The initial pressure of the gas mixture in atmospheres (e.g. for a pressure of 40 Torr,
PRES 0.053)
REAC – Moles of reactant in initial mixture (e.g. REAC H2 1.0). The mole fractions of reactants are normalised so the absolute magnitudes of the input mole quantities are unimportant.
The total length of time (seconds) for which the reaction is allowed to proceed is specified using the keyword TIME (e.g. TIME 5). The time interval (seconds) for solution printouts to the printed output file (tign.out) is specified by the DELT keyword (e.g. DELT 1.E-4).
A sample SENKIN input file is given below. The gas temperature is fixed at 2000 K and the pressure is fixed at 0.053 atm (40 Torr). The reaction time is 5 sec with printouts every 1×10-4 sec, and the reacting mixture is 1% CH4/H2.
CONT
PRES 0.053
TEMP 2000
TIME 5.0
DELT 1.E-4
REAC H2 0.99
REAC CH4 0.01
END
4.8.2. Running the SENKIN code (senk.exe)
The Chemkin
Interpreter must be run (with no errors in the linking file) prior to the
SENKIN code being executed. Once this
has been done there is no need to repeat this step (unless a change in the
reaction mechanism, chem.inp, has been made). A Senkin input file (senk.inp) can be written on a PC (using the
notebook package) before being transferred onto grendel by a FTP program or
within the user directory on grendel using pico (typing pico senk.inp will
create a file called senk.inp which can then be edited). SENKIN is run by typing senk <senk.inp >senk.out.
4.8.3. SENKIN Printed Output File (tign.out)
This file states what type of problem was investigated (e.g. “temperature is held constant”), the initial conditions (i.e. pressure and temperature), initial species mole fractions and the result of each time integration. A shortened output file is shown below.
SENKIN: Sensitity Analysis
Author: Andy Lutz
CHEMKIN-II Version 3.0, March 1994
DOUBLE PRECISION
Sensitivity analysis will not be performed.
Temperature is held constant.
Initial Conditions:
Pressure (atm) = 5.3000E-02
Temperature (K) =
2.0000E+03
Density (gm/cc) =
6.9633E-07
Mole Fractions:
H2 = 9.9000E-01
H = 0.0000E+00
C = 0.0000E+00
CH = 0.0000E+00
CH2 = 0.0000E+00
CH3 = 0.0000E+00
CH4 = 1.0000E-02
C2H = 0.0000E+00
C2H2 = 0.0000E+00
C2H3 = 0.0000E+00
C2H4 = 0.0000E+00
C2H5 = 0.0000E+00
C2H6 = 0.0000E+00
Time Integration:
t(sec)= 0.0000E+00
P(atm)= 5.3000E-02 T(K)=
2.0000E+03
H2 = 9.90E-01 H
= 0.00E+00 C = 0.00E+00
CH = 0.00E+00 CH2
= 0.00E+00 CH3 = 0.00E+00
CH4 = 1.00E-02 C2H
= 0.00E+00 C2H2 = 0.00E+00
C2H3 = 0.00E+00 C2H4
= 0.00E+00 C2H5 = 0.00E+00
C2H6 = 0.00E+00
t(sec)= 1.0002E-04
P(atm)= 5.3000E-02 T(K)=
2.0000E+03
H2 = 9.90E-01 H
= 8.47E-05 C = 5.49E-15
CH = 7.70E-13 CH2
= 3.12E-09 CH3 = 2.36E-05
CH4 = 9.98E-03 C2H
= 2.52E-17 C2H2 = 4.84E-11
C2H3 = 1.88E-12 C2H4
= 4.01E-09 C2H5 = 2.71E-10
C2H6 = 4.37E-10
(Intermediate datasets omitted for brevity)
t(sec)= 5.0000E+00 P(atm)= 5.3000E-02 T(K)= 2.0000E+03
H2 = 9.88E-01 H
= 6.98E-03 C = 1.35E-09
CH = 2.06E-09 CH2
= 1.00E-07 CH3 = 8.36E-06
CH4 = 4.80E-05 C2H
= 2.12E-07 C2H2 = 4.90E-03
C2H3 = 1.02E-07 C2H4
= 1.58E-06 C2H5 = 5.52E-11
C2H6 = 4.14E-11
Binary file has 1054
time datasets.
The final dataset in the output file gives the species mole fractions resulting from the input species reacting (as per the chem.inp reaction mechanism) for the specified time. The output file (tign.out) can be examined using Pico, or tign.out can be copied over to a PC (using SSH Secure File Transfer Client) and opened using Word.
4.9. Limitations of SENKIN
The SENKIN results (Chapters 5-7) presented within the present work were produced using a chosen input gas mixture, at a fixed gas temperature and pressure (problem type D). Limitations inherent in this approach include the following.
1. The assumption of a single temperature for all reactions in a given simulation. This is clearly an approximation in the case of hot filament (HF) activated gas mixtures which involve large temperature gradients [[26]] but is a more reasonable assumption in the case of low power (< 3 kW) microwave plasma activation.
2. No electron impact dissociation, ionic reactions or surface chemistry are included in the modelling. Reaction is thus initiated solely by thermal dissociation.
3. No transport into or out of the reaction volume is considered. To mimic experiment, therefore, it is necessary to run the simulation for a finite (user selected) time, t. The calculations reported here employ t = 5 s.
4.10. Automatic looping of SENKIN
If a range of initial conditions (e.g. temperature, pressure or gas mixture) are required, SENKIN requires the user to successively edit senk.inp, run SENKIN and examine the output file tign.out. The program cycle was written [[27]] in order to run SENKIN repeatedly (while changing the proportion of the two input species, CO2 and CH4) automatically.
The user sets the range of CH4 mole input (e.g. 0 – 100%), keyword data such as initial conditions (pressure and temperature) and integration commands (total time and dataset separation) within the program. Running the program then carries out the following tasks:
- opens a senkin input file (senkp.inp),
- writes the keyword data information into it (with the species moles input at the initial values set),
- runs SENKIN,
- redirects the input species moles and the last dataset into a separate file,
- increases the input moles of CH4 and
- the process is repeated until the full range of input moles of CH4 has been covered.
4.10.1. The cycle Program
The program code is explained below, with added comments (not part of the code) added in bold type. These statements have been prefixed by ! therefore the following text can be copied into a notebook file and run as a program.
#!/usr/bin/perl
my $I, $conc1, $conc2; !Defines the variables conc1 and
conc2
my $rc;
for ($I=0; $I<=100000;
$I = $I+1000) {
!Sets the input mole range
for CH4 (0-100%) and % steps (1%).
$conc1 = $i/10000;
$conc2 = (10000-$I)/10000;
!sets %CO2 = 100% - %CH4
open (SENKIN, “>senkp.inp”) or die “Can’t open senkp.inp:
$!”;
!opens a file called
senkp.inp
print SENKIN “CONT\n”;
print SENKIN “PRES 0.053\n”;
print SENKIN “TEMP 2000\n”;
print SENKIN “TIME 5.0\n”;
print SENKIN “DELT 1.E-4\n”;
!input keywords are
written into senkp.inp
printf SENKIN “REAC CH4 %8.5f\n”, $conc1;
!CH4 input moles written
into senkp.inp
printf SENKIN “REAC CO2 %8.5f\n”, $conc2;
!CO2 input
moles written into senkp.inp
print SENKIN “END”;
close SENKIN;
printf STDERR “%8.5f\n”, $conc1; !%CH4 printed on screen
$rc = 0xffff & system “senk <senkp.inp
>senkp.out”; !SENKIN is run
if ($rc != 0) { die “senk did not run:$rc”; }
open (SENKOUT, “tail –12 tign.out|”) or die “Can’t open tign.out: $!”;
!Last 12 lines of tign.out
are read (containing last dataset)
my $line = “”;
while (<SENKOUT>) {
chomp;
if ( /=/ ) { $line = $line . $_; }
}
close SENKOUT;
printf “%8.5f %8.5f “, $conc1, $conc2;
!%CH4 and %CO2 are written
to screen (or redirected to results file, see below)
print $line, “\n”; !last
dataset is written as with last line
}
Note: the user can define all the input parameters such as pressure, temperature, input species, etc. by editing the cycle program (e.g. by using Pico).
4.10.2 Running cycle
Cycle is run by typing ./cycle >results. This command runs the program and also redirects the output to a file called results. The input moles of CH4 are displayed on screen each time SENKIN is run (so the progress of the program can be followed). The output file from cycle (the file results in this case) is formatted with a line for each gas mixture which also contained the output mole fractions for all the species defined in the chem.inp file, i.e.
Input moles CH4 Input moles CO2 Species name = species mole fraction Species name etc…
0.24000 0.76000
H2 = 1.33E-01 H
= 2.56E-03 O (etc…)
0.26000 0.74000
H2 = 1.58E-01 H
= 2.79E-03 O (etc…)
This output file can then be copied over to a PC and imported into the
Excel spreadsheet package thus allowing
manipulation and presentation of the data.
Thus, plots of species mole fraction versus gas mixture can be
produced. The user can modify cycle
to suit the problem being
investigated. Two variations are the
programs Temp and Time which
run for a range of temperatures and times respectively. These are given in Appendix 5.