Section 1
Chapter 1 : Introduction
Since its
first documented mining in India around the fourth century B.C. diamond has
been highly regarded for its brilliance.
In a second century B.C. manuscript it was reported that the Chinese had
realised diamond’s “industrial“ use stemming from its unequalled hardness and
were importing diamonds from India.
Diamond’s
hardness has in turn lent itself to its own name. The name diamond is derived from the Greek adamaz (adamas),
which is translated as ‘unconquerable’, and while used originally to refer to
any hardened material such as steel, now refers solely to diamond.
It is not
surprising then that today diamond is sought after as a jewel and is used in a
wide range of industrial applications, for example, grinding, polishing, wear
resistant coatings, etc.
This chapter
opens with a brief summary of the structure and properties of diamond, and of
synthetic routes to diamond, based on high pressure high temperature (HPHT)
methods and the more recent chemical vapour deposition (CVD) route. The complex chemistry underpinning the CVD
methods of growth is also discussed, as are the present and possible future
applications of synthetic diamond.
1.1 Structure and Properties of Diamond
Diamond is
simply a 3D network of tetrahedrally bonded sp3 hybridised carbon
atoms having a cubic-close packed crystal structure analogous to that of zinc
blende. Each carbon atom is s-bonded to
four neighbouring carbon atoms. This
differs from the structure of graphite in which a hexagonal arrangement of s-bonded sp2
hybridised carbon atoms exist in 2D sheets, where each sheet is weakly bonded
to its neighbour. This structure
attributes itself to the softness of graphite.
These two structures are shown in figure 1.1.
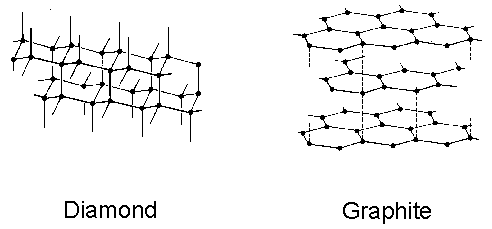
Figure 1.1 The lattice structures of diamond and graphite. Carbon atoms are noted by the black spheres, s-bonds by the solid lines and weak inter-sheet bonds by the dashed lines.
While diamond,
like graphite, is an allotrope of carbon its scarcity adds to its
mystique. This scarcity, relative to
that of graphite, is a facet of a large activation energy preventing thermal
conversion from graphite to diamond.
However, the enthalpy of formation of diamond is strangely close to that
of graphite, being only 2.9 kJ mol-1 higher than that of diamond
(measured at 298 K and 1 atm). Diamond,
being the denser allotrope of carbon, (rdiamond = 3513 kg m-3: rgraphite = 2260 kg m-3
at 293 K) is the most stable at high pressures.
Diamond is often referred to as the ultimate engineering material. Its extreme properties described here make it an attractive engineering material for a large number of applications, some of the most common uses and applications of diamond are described in detail later in this chapter.
Diamond’s
structure of strong covalent bonds holding a tetrahedral network of carbon
atoms makes it the densest and hardest of any known material. The elastic modulus and compressibility of
diamond are also unsurpassed as a facet of its structure; it is robust and
resistant to both radiation damage and chemical corrosion.
While diamond
has a number of extreme physical characteristics, its thermal and electrical
properties are also of interest.
Natural diamond is an insulator with a wide band gap of 5.49 eV. By altering the diamond lattice, with the
inclusion of atoms with valences different to that of carbon, it is possible to
form both n- and p-type doped diamond.
Uncharacteristic
to non-metal materials, diamond has an unparalleled thermal conductivity, four
times greater than that of copper at room temperature. Metals are generally good thermal and
electrical conductors due to the high mobility of electrons within the lattice. Undoped diamond having a covalent lattice
has very little electron mobility and is therefore an electrical
insulator. However, due to the
extremely high degree of rigidity within the structure crystal vibrations
(phonons) allow high thermal conductivity.
Pure diamond
crystals have a very wide optical transparency, ranging from
0.22 mm, in the
ultraviolet, to the far infrared. Due
to its high symmetry, very few intrinsic absorbance bands are present,
impurities (typically nitrogen) in the lattice represent the main source of
absorbance bands. However, lattice
defects reveal optical activity of the electronic and vibrational transitions.
Table 1.1 summarises some of the unique properties of diamond.
|
Property |
Value |
Note |
|
Vickers
Hardness (kg mm-2) |
12000 – 15000 |
Hardest
known material |
|
Compressibility
(m2 N-1) |
8.3 ´10-3 |
Lowest
known compressibility |
|
Elastic
Modulus (N m-2) |
1.2 ´ 1012 |
Highest
elastic modulus known |
|
Thermal
conductivity (W cm-1 K-1)a |
2 ´ 103 |
Highest
known thermal conductivity |
|
Thermal
expansion coefficient (K-1)a |
0.8 ´ 10-6 |
Close
to Si (0.57 ´
10-6) |
|
Electrical
resistivity (W
m) |
1016 |
High
resistivity |
|
Doped
electrical resistivity (W m) |
0.1 - 104 |
|
|
Chemically
and biologically inert |
||
|
Optically transparent from deep UV to
Far Infrared |
||
|
High resistance to radiation damage |
||
|
a measured at
room temperature |
||
Table 1.1 Intrinsic properties of natural diamond.
1.2 Synthesis of Diamond
Since
Tennant’s realisation in 1776 that diamond is solely a form of carbon[1],
and the discovery in the 19th century that diamond is formed deep in
the earth’s crust, many attempts were made to simulate the high pressures and
temperatures required using graphite as a carbon source. It was not until 1954 that the synthesis of
diamond by a High Pressure High Temperature (HPHT) technique by the General Electric
Company was successful[2].
While the
production of HPHT diamond and the mining of diamond were providing industrial
diamonds predominately for the cutting industry, a new technique for growing
diamond at low pressures was also discovered between 1953 and 1954 [3],[4]. This new technique involved the Chemical
Vapour Deposition (CVD) of diamond from a carbon containing gas precursor at
low and atmospheric pressures. While a
number of different growth techniques have been demonstrated the same principle
is followed. Growth is carried out by
the deposition of carbon from an activated gas-phase hydrocarbon onto a
suitable substrate material maintained at a temperature of between 1000 and
1400 K. The major differences between
the different techniques used are mainly related to how the gas-phase
environment is activated. A description
of the most common reactor types follows later in this chapter.
1.3 HPHT synthesis of Diamond
As mentioned
previously, diamond being the densest allotrope of carbon is the most stable
form at high pressures. This fact,
together with the knowledge that diamond is formed at high temperatures within
the earth’s crust, stimulated research into a HPHT technique for the synthesis
of diamond. The technique, first
published in 1954, is still used in the production of industrial grade
diamond. The basis of the formation is
the crystallisation of diamond from a melt of metal-solvated carbon at
pressures of between 50 and 100 kBar and temperatures ~1800 - 2300 K.
While HPHT
techniques have so far been used in the synthesis of industrial grade diamond,
recent reports by the General Electric Company have confirmed use of a HPHT
technique to manipulate the colour centres within natural diamonds for the gem
market[5].
Unfortunately,
HPHT synthesis of diamond suffers from high cost and the inability to produce
coatings or films of diamond. The CVD
of diamond therefore has an advantage over HPHT synthesis in that the
deposition of diamond is much more flexible, being governed by the design of
the reactor.
1.4 Diamond synthesis by Chemical Vapour
Deposition (CVD)
The deposition
of crystalline diamond from a carbon containing gas mixture was first
demonstrated at about the same time as the development of HPHT techniques in
the mid-1950’s. The first attempts at
diamond CVD at pressures less than an atmosphere resulted in extremely low
deposition rates and were restricted to growth onto diamond substrates. Patent applications by Eversole[6]
of the Union Carbide Corporation in 1962 outlined the low pressure CVD
technique used, this was far superior to earlier techniques that also produced
large quantities of graphitic carbon.
Angus et al.3 improved
Eversole’s technique by discovering that atomic hydrogen preferentially etches
deposited graphite over diamond. It was
not until 1981, however, that Spitsyn et al.[7]
succeeded in depositing diamond onto substrates other than diamond at growth
rates of approximately 1mm hr-1. During the early 1980’s growth rates were
improved by a number of research groups and companies by studying various
growth techniques to optimise the diamond deposition conditions[8],[9].
The problem
associated with understanding how diamond CVD occurs is one of appreciating
under what conditions and limitations a metastable material can be grown. This encompasses a large number of problems
in itself, including the nature of the complex gas-phase reactions, and the
surface and bulk chemistries of diamond formation.
Increased
understanding of the CVD process in terms of the activation of the gas-phase
environment, the transport of species, and the growth process has fuelled
development of the procedure. The
process of diamond CVD, irrespective of the method of production, may be
described in a number of separate yet interlinked stages. The reactants, typically a dilute
hydrocarbon / hydrogen gas mixture, are activated forming the important radical
species and atomic hydrogen. The
transport of the species to the growing surface is the next consideration; this
may occur by convection or by diffusion and is determined by the technique
used. At the growing surface, the
gas-surface interactions initiate the necessary surface chemistry. Diamond growth is possible if the substrate
temperature is maintained at between 1000 and
1300 K. This process is summarised in figure 1.2.
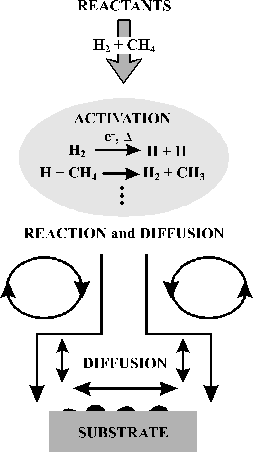
Figure 1.2 Schematic representation of the diamond CVD scheme adapted from a review article by Goodwin and Butler[10]
In the next
chapters the gas-phase and surface chemistry will be discussed in terms of a
low power CVD reactor. Such reactors
have been studied in more detail than DC-arcjet systems where high reactant
flux and gas temperatures may present subtle differences in the relative mole
fractions and transport of species. The
discussion will also be limited to low-pressure reactors, however recent
results from high-pressure (above one atmosphere) CVD systems have demonstrated
very high growth rate and quality deposition[11].
1.5 Gas-phase chemistry
Diamond CVD is
most commonly carried out with a reactant gas mixture of approximately 1-2%
methane in hydrogen. Although it is
known that diamond growth is relatively unaffected by the carbon source gas
used[12],[13],
methane or acetylene are most commonly employed. In order to initiate reactions in the gas-phase it is necessary
to activate the gases, this may be carried out by a number of different methods
and defines the technique used. While
the different CVD methods will be discussed in detail later in this chapter,
the activation of the gas-phase reactants may involve thermal activation (e.g.
a hot filament), use of an electrical discharge (e.g. microwave, DC or RF) or
by flame (e.g. oxyacetylene torch). The
activation of the gas mixture, irrespective of the method used, produces atomic
hydrogen. Atomic hydrogen is the
driving force behind the gas-phase and surface reactions and is clearly one of
the most important species present in terms of diamond film growth and quality.
The majority
of studies of the gas-phase and growth process have been carried out in a hot
filament system (see Chapter 1.8.1) and, while a number of factors discussed
here may be technique-dependant, it is thought that, in general, the results
obtained may be applied to diamond deposition as a whole.
1.5.1 Atomic hydrogen production
In hot
filament systems, atomic hydrogen (from herein also referred to as H atoms) is
produced heterogeneously on a hot filament surface via thermal decomposition,
shown in reaction 1.1.
H2 ® 2H Reaction
1.1
The H atoms
diffuse rapidly into the reactor. The
atomic hydrogen concentration, [H], at the filament surface scales
approximately as would be expected for thermal equilibrium. However, measurements of the atomic hydrogen
absolute concentration show that only between 12%[14]
and 60%[15]
of the expected [H] exists for a given filament surface area, filament
material, reactor pressure and hydrocarbon content. This has been attributed to the rapid diffusion of atomic
hydrogen from the filament surface to cooler regions of the reaction
chamber. For this reason, in the cooler
regions of the reactor a super-equilibrium of atomic hydrogen exists. Process pressures, typically being below 1
atmosphere, also maintain the atomic hydrogen super-equilibrium by causing atomic
hydrogen recombination in the gas phase to be a slow process.
The production of atomic hydrogen within plasma-enhanced reactors (such as microwave and DC-arc) is achieved via direct coupling of energy into the free electrons within the plasma. This produces H atoms homogenously via reaction 1.2 shown.
H2 + e- ®
H + H + e- Reaction 1.2
This reaction
occurs through successive vibrational excitation of H2 by electron
impact.
It is worth
noting that in regimes where the pressure exceeds 100 Torr it is thought that
through inelastic collisions electrons may be able to transfer energy to heavy
particles. This will result in heating
of the gas, leading to H2 dissociation by impact with heavy
particles, M, via the general reaction 1.3
H2 + M ® 2H + M Reaction 1.3
At these
temperatures (~ 3000 K) the
production and loss rates of H atoms are such that an equilibrium value may be
reached for the local gas temperature.
Plasma-enhanced
reactors have a distinct advantage over hot filament systems in terms of H atom
production, as the active surface area of the filament restricts the production
of H atoms. It has long been understood
that under conditions of high gas-phase carbon content the filament surface
becomes poisoned. This occurs due to
the growth of a graphitic layer on the filament from a gas-phase environment,
in which the carbon content surpasses the solubility limit in hydrogen. The solubility limit is a function of the
filament temperature and inhibits the catalytic effect of the filament.
1.5.2 Atomic hydrogen loss mechanisms
The production
and loss rates of atomic hydrogen in a reactor are such that a steady state
regime exists. The destruction of H
atoms, assuming no atomic hydrogen leaves the reaction chamber, occurs mainly
via either homogenous or wall recombination mechanisms.
The homogenous
recombination of atomic hydrogen, under typical conditions prevailing in a CVD
reactor, occurs at a slow rate. This
further allows the super-equilibrium of atomic hydrogen to exist at low local
temperatures, thus supporting the transport of H atoms to the growing surface
or the reactor chamber walls. The
homogenous recombination occurs via reaction 1.4,
H +
H + M ® H2 + M Reaction
1.4
where M is a
third body, facilitating the removal of excess heat of recombination. This reaction rate is pressure dependant
with a characteristic reaction time at a pressure of 20 Torr of the order of 1
s [16],[17].
The presence of a small quantity of hydrocarbon, typically methane, presents a removal route for H atoms via the gas-phase reactions.
H + CH3 +
M ® CH4 + M Reaction 1.5
H + CH4 ® CH3 + H2 Reaction
1.6
These two
reactions compete with reaction 1.4 and hence determine the recombination rate
of atomic hydrogen.
The most
important route for the loss of H atoms is heterogeneous recombination. This may occur either as the atomic hydrogen
impinges on the reactor walls, or due to collision with the growing diamond
surface. Studies have shown the growing
diamond surface acting as an H atom ‘sink’, the concentration profile at the
substrate surface being a function of the substrate temperature13. The loss of H atoms to a diamond surface may
be expressed in term of the recombination coefficient, gH, which is
defined as the atomic hydrogen loss rate at the surface divided by the H atom
collision rate with the surface.
Experimental[18]
and theoretical[19]
measurements of this factor have all been in agreement. Krasnoperov et al.[20]
measured gH over a wide
substrate temperature range, establishing the functional form shown in figure
1.3.
![]()
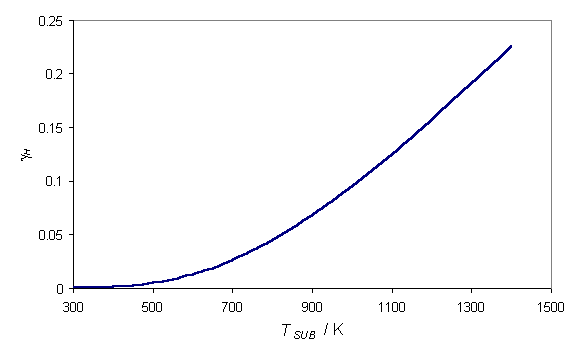 Figure
1.3 Graph showing the recombination
coefficient of H at the diamond surface as a function of Tsub
adapted from reference 10.
Figure
1.3 Graph showing the recombination
coefficient of H at the diamond surface as a function of Tsub
adapted from reference 10.
At substrate temperatures, typically used during diamond CVD (~1000-1300 K), the recombination coefficient is high, signifying that the surface is acting as a sink for H atoms. The recombination reaction,
2H ® H2 Reaction 1.7
is exothermic
(104 kcal mol-1) and in many reactor systems is the major
contributor to heating the substrate.
1.5.3 Hydrocarbon Gas-phase Chemistry
The role of
atomic hydrogen is also one of ‘fuelling’ the hydrocarbon gas-phase
chemistry. In the gas-phase, atomic
hydrogen participates in a number of abstraction reactions of the hydrocarbon
reactant and its ‘abstracted’ species.
Figure 1.4 summarises the principal production and loss routes of carbon
species in a CVD reactor operating with a CH4/H2 gas
mixture.
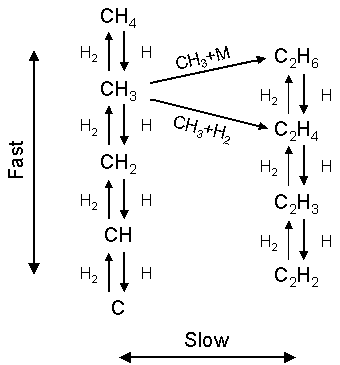
Figure 1.4 Principal C1 and C2 gas-phase reaction scheme illustrating the fast hydrogen shift reactions and the slower bimolecular C1 to C2 hydrocarbon forming steps. Adapted from reference [21].
It is worth
noting that this reaction scheme only deals with C1 and C2
species. While C3 and higher
species are present in the gas-phase, it is thought that the relative concentrations
of these species are sufficiently low so as not to influence the reaction
scheme shown[22]. Hydrogen and atomic hydrogen, being the
major gas-phase species present in the reactor, dominate the reactions, with
the hydrogen transfer reaction rates being generally greater than those
describing the bimolecular hydrocarbon reaction rates.
The series of
interconnected hydrogen shift reactions (left-hand column of figure 1.4) occur
rapidly in both the forward and reverse directions due to low activation
energies. The rate of reaction between
atomic hydrogen and any carbon-containing species will obviously be a function
of the local gas temperature and the local atomic hydrogen concentration. This relationship defines the production and
distribution of C1 species throughout the reactor.
Due to the
complex nature of the reaction scheme and the gas temperature gradients that
exist within a CVD reactor, studies of the gas-phase environment are deemed
necessary to improve our understanding of this system. The first hydrocarbon concentration
measurements of the diamond CVD environment were carried out by Celii et al.[23]
using an infrared diode laser absorption technique to detect acetylene, methyl
radicals and ethylene in a hot filament reactor.
This study found
that high concentrations of CH4 and C2H2
existed together with low concentrations of CH3 and C2H4
from an input CH4 / H2 gas mixture. Bearing in mind that the absorption
technique used sampled a column of inhomogeneous gas, a rotational temperature
of ~600 K was
obtained from C2H2 detection. This reflects an average gas-phase temperature ranging from the
hot filament to the cold reactor walls.
Simple
gas-phase equilibrium calculations as in figure 1.5 for a 1% CH4 / H2
gas mixture, clearly show that the conversion of CH4 to C2H2
occurs at gas temperatures that exist during diamond deposition.
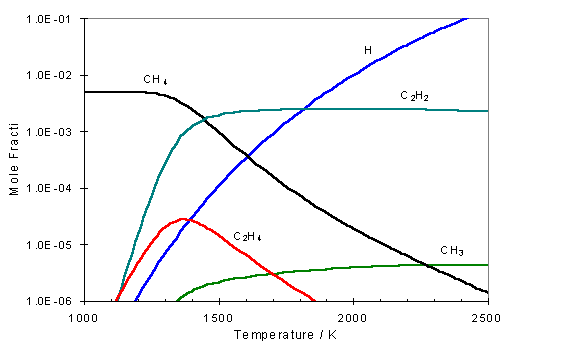
Figure 1.5 Gas-phase equilibrium calculation carried out using the CHEMKIN computer package[24] for a 1% CH4/H2 gas mixture using GRI-MECH 3.1[25] reaction rate constants
The study
concluded that a significant fraction of the CH4 is converted into C2H2
at high gas temperatures within the reactor.
The concentrations of acetylene measured in this study represented a
10-20% conversion from methane. Since
this conversion occurs via a number of sequential reactions each involving
atomic hydrogen, it was recognised that a complex gas-phase chemistry scheme
must exist within the CVD reactor.
Subsequent studies of the gas-phase chemistry in both hot
filament and microwave systems by a number of detection methods have given an
indication of the gas composition. One
interesting feature resulting from these studies is that the mole fractions of
carbon containing species in the activated gas are essentially independent of the
input carbon-containing precursor.
Molecular-Beam Mass Spectrometry (MBMS) studies carried out by Tsang et al.[26]
have shown this independence for input gas mixtures of CH4/H2, C2H4/H2
and C2H2/H2 in a hot filament reactor as a
function of filament temperature (for a filament temperature > 2200 K). The results of this study are shown in
figure 1.6.
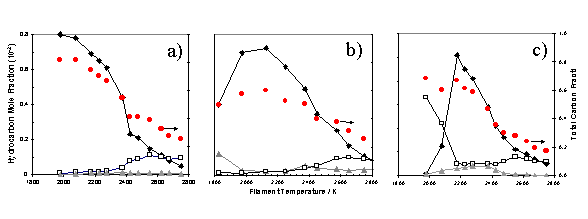 Figure
1.6 MBMS results showing the mass
fractions of stable species as a function of filament temperature, measured 6mm
from the filament surface in a hot filament CVD reactor operating on gas
mixtures of a) 1% CH4 / H2 b) 0.5% C2H4
/ H2 and c) 0.5% C2H2 / H2. Symbols
shown relate to CH4 (¨), C2H2 (o) and C2H4
(p). The
carbon balance (l) is shown to decrease with increasing filament temperature being a consequence
of the Soret effect.
Figure
1.6 MBMS results showing the mass
fractions of stable species as a function of filament temperature, measured 6mm
from the filament surface in a hot filament CVD reactor operating on gas
mixtures of a) 1% CH4 / H2 b) 0.5% C2H4
/ H2 and c) 0.5% C2H2 / H2. Symbols
shown relate to CH4 (¨), C2H2 (o) and C2H4
(p). The
carbon balance (l) is shown to decrease with increasing filament temperature being a consequence
of the Soret effect.
The conversion
of a C1 species to a C2 species is carried out via rapid
H atom abstraction. If, for example,
methane is introduced into a reaction scheme whereby the H/H2
fraction is below a few percent, methyl radicals (CH3) may be formed
via reaction 1.8
H + CH4 ¾ CH3 + H2 Reaction 1.8
Recombination
of these methyl radicals will go to form C2 species via the
reactions,
CH3 + CH3 ¾ C2H6 Reaction 1.9
CH3 + CH3 ¾ C2H5 + H Reaction 1.10
CH3 + CH3 ¾ C2H4 + H2 Reaction 1.11
Once the C2
species is formed it is rapidly converted to the thermodynamically favoured
stable species C2H2 via the reaction scheme:
C2H6 +
H ®
C2H5
+ H2 Reaction 1.12
C2H5 +
H ®
C2H4
+ H2 Reaction 1.13
C2H5 +
M ®
C2H4
+ H + M Reaction 1.14
C2H4 +
H ®
C2H3
+ H2 Reaction 1.15
C2H3 +
H ®
C2H2
+ H2 Reaction 1.16
C2H3 +
M ®
C2H2
+ H + M Reaction 1.17
Modelling
studies, like those used in deriving figure 1.5, indicate that no analogous
gas-phase process exists that allows the conversion of acetylene, a C2
species into a C1 species.
This is clearly in disagreement with experimental studies and has
prompted some to postulate that the conversion may occur heterogeneously on the
reactor wall surface10.
Since a portion of this thesis deals with experimental studies into this
problem, further discussion will be presented in chapter 3.
From modelling
studies carried out by Dandy and Coltrin[27],
it is clear that CH3, the methyl radical, is the most abundant C1
radical species present in the gas-phase, under typical low power CVD process
conditions. Methyl radicals are formed,
via reaction 1.8. While numerical
simulations show this reaction to be in partial equilibrium at distances
greater than 1 mm from the substrate surface, close to the surface the
comparative lack of H atoms, due to surface recombination, causes the partial
equilibrium to collapse.
Mass
spectroscopy studies carried out by Harris et al.22, using a
quartz sampling tube, positioned at the surface of either silicon or platinum
substrates, have shown that CH4 and C2H2 are
the main species present at a growing surface.
The analysis of the gas-phase composition confirms a non-equilibrium
nature at the substrate surface. The
concentrations of species such as CH4, C2H2, CH3
and C2H4 were measured and found to be sufficient to
account for diamond deposition. Larger
hydrocarbons (i.e. C3 and greater) were present in trace quantities
only and therefore deemed unimportant for diamond growth.
Resonance
Enhanced Multiphoton Ionisation (REMPI) techniques (described in detail in
appendix 1) have been used in a number of studies for the detection of H and CH3,
both thought to be important species in the diamond growth environment in a hot
filament CVD reactor. Early REMPI
studies[28]
showed how the filament temperature influences the H atom production. The studies concluded that the atomic
hydrogen concentration increases monotonically with increasing filament
temperature. For gas mixtures with high
CH4/H2 fractions the increase is less pronounced, this
can be explained by carbon deposition on the filament surface10.
1.5.4 Influence of
trace non-hydrocarbon additions on gas-phase chemistry
Several
studies have reported enhanced growth rates and / or quality due to the
addition of trace amounts of non-hydrocarbon gases (such as oxygen[29],[30],
nitrogen[31]
and halogens[32]) into a
diamond-depositing reactor.
Reports of
enhanced growth as a result of adding oxygen to a hydrocarbon gas mixture have
predominantly focused on microwave based CVD systems[33],
similar effects have been demonstrated in a DC-arcjet reactor[34]. Comparable experimental studies have also
been carried out in a hot filament system[35],
but it is worth noting that even trace quantities of oxygen will oxidise and
ultimately destroy a filament.
Since the
complex carbon-oxygen-hydrogen chemistry involved is beyond the scope of this
thesis only a brief outline will be given here. The gas-phase production of CO via oxidation of acetylene as
shown in reaction 1.18 is highly exothermic with a free energy of reaction (DG0) = -136.6
kcal mol-1 at 1000K.
C2H2
+ O2 ¾ 2CO
+ H2 Reaction 1.18
This reaction
together with the analogous reaction 1.19,
2CH4 + O2 ¾ 2CO
+ 4H2 Reaction 1.19
which also
possesses a large negative DG0, dominate the
carbon-oxygen chemistry as at gas-phase temperatures typical within CVD
reactors the equilibria of both above reactions lies far to the right.
By analysing
published experimental data Bachmann et al.[36]
were able to characterise a regime in which diamond deposition occurs within a
narrow range of C/H/O gas compositions.
The Bachmann triangle (also known as the C/H/O diagram) identifies a
tie-line in the ternary system corresponding to high quality growth at high
growth rates. The tie-line points to a
link between the growth mechanism and a 1:1 carbon/oxygen ratio in the input
gas mixture.
The influence
of the addition of nitrogen (and other nitrogen containing species, such as NH3)
into a carbon-hydrogen gas mixture at gas temperatures over 600 K is complex
and will be investigated and discussed in detail in Chapters 4 and 6 in
reference to studies in Hot filament and DC-arc CVD reactors.
1.6 Surface Growth of Diamond
While atomic hydrogen is the main initiator of the gas-phase chemistry it is also crucial at the growing surface. With hydrogen and atomic hydrogen being the major constituents of the gas-phase environment the diamond surface will be mainly hydrogenated. The surface will therefore have a fraction of open non-hydrogenated sites, which are regulated by the dynamic equilibrium between the two surface reactions,
CDH +
H ®
CD* + H2 Reaction 1.20
CD* +
H ®
CDH Reaction
1.21
where CDH
signifies a hydrogen terminated surface site and CD* a
non-terminated surface site. The
thermal desorption of H from the diamond surface (the reverse of reaction 1.21)
may be ignored as the C-H bond strength is high. Studies by Goodwin[37]
have shown that the formation of hydrogen terminated surface sites occurs
principally as a result of reaction 1.21 rather than the reverse of reaction
1.20, for H atom mole fractions greater than 10-4. Such a conclusion is based on the fact that,
under typical CVD conditions, the H atom mole fraction at the substrate surface
is generally greater than 10-3.
A review
article by Goodwin and Butler10 shows that by balancing the CD*
production and loss rates from reactions 1.20 and 1.21, a value of the fraction
of open sites, f, may be
obtained using equation 1.1,
![]() Equation
1.1
Equation
1.1
where g1 is the
abstraction probability via reaction 1.20, and g2, is the
adsorption probability from reaction 1.21.
Both graphite
and diamond will, to some extent, be co-deposited within a CVD
environment. Angus et al.[38]
showed that atomic hydrogen preferentially etches graphite approximately 50
times faster than diamond at typical growth temperatures, explaining why high
purity CVD diamond is obtained by using high H:C input gas ratios.
As already
mentioned, atomic hydrogen terminates the dangling-bonds present at the growing
surface. This is important since by
terminating the surface the reconstruction of diamond to graphite is
inhibited. The surface termination is
also believed to enhance nucleation.
Growth
mechanisms have been proposed for many hydrocarbon species. Growth onto the various surfaces of diamond,
typically defined by their Miller indices, has been shown to occur at growth
rates based on the lattice energy and the steric hindrance during addition. Semi-empirical quantum mechanical calculations
have been carried out to measure the energy path of diamond growth onto a
hydrogenated (111) surface[39]. The lowest energy scheme was shown to be
initiated by the monolayer (111) surface coverage by methyl groups, which
consequently forms a lattice via the incorporation of a methyl radical or
cation. However, it has since been
shown that steric effects between the surface terminating methyl groups prevent
this scheme[40].
Further growth
studies concentrating on C1 species addition to the (111) surface
have concluded that similar steric difficulties exist. The addition of C1 species onto a
diamond surface was shown to be ‘optimised’ for the hydrogenated (100) surface
where only one carbon is required to form two bonds to the surface. The hydrogenated (100) surface has been
shown theoretically to exist in two forms, the dihydride surface, denoted
(100)-(1x1):2H, and the monohydride surface, (100)-(2x1):1H. However, the dihydride surface consisting of
closely neighbouring hydrogen atoms is subject to huge steric strain. This has since been proven by studying grown
(100) CVD films using Scanning Tunnelling Microscopy and Atomic Force
Microscopy[41], and
successive low-energy electron diffraction and high-resolution electron energy
loss measurements[42].
Garrison et
al.[43]
and Huang and Frenklach[44]
have proposed growth schemes based on the addition of methyl radicals onto a
monohydride (100) surface. The Garrison
mechanism outlined in figure 1.7 proceeds via an initial surface hydrogen
abstraction followed by the addition of a methyl radical.
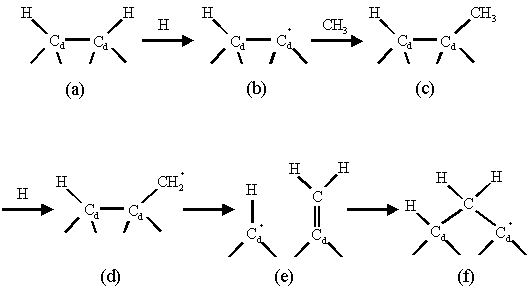
Figure 1.7 The Garrison mechanism for dimer opening and carbon insertion allowing growth to the (100) surface. This mechanism was adapted from reference 10.
Further hydrogen abstraction occurs from the methyl group forming a primary intermediate for a b-scission reaction. b-scission reactions are thought to play an important part in the etching of non-diamond carbon and in promoting the growth of single atomic diamond layers. Figure 1.8 schematically illustrates a b-scission reaction mechanism occurring at a surface methyl group.
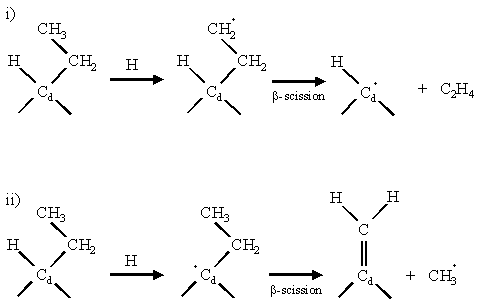
Figure 1.8 b-scission reaction
mechanisms adapted from reference [45]. Two possible ways to attack a surface-bonded
ethyl group are shown. An H atom is
first abstracted by gas-phase H to create a radical site. In scheme (i) a methyl hydrogen is
abstracted, whereas in scheme (ii) a hydrogen on the diamond surface carbon (Cd)
is abstracted. Both schemes cause the b-carbon bond to break,
allowing double bond formation to the carbon from which the hydrogen was
abstracted.
The radical
site and the methylidene intermediate subsequently react to develop a bridging
carbon.
The mechanism
proposed by Huang and Frenklach differs only in that the final stage is reached
via a triangular intermediate stage bypassing the b-scission
reaction, however an activation energy for the formation of the intermediate
state of ~230 kJ mol-1 suggests
the b-scission
route is more favourable.
While the
Garrison mechanism is now generally accepted for growth onto a (100) surface
various adaptations have been proposed.
Harris and Goodwin[46]
adapted the mechanism to take into account bridging between dimers after
insertion. Thermochemical analysis of
this process revealed that, while the distance between two adjacent dimer
carbon atoms is too large to be bridged by the methylidene intermediate,
successive addition via the Garrison mechanism significantly reduces the
carbon-carbon distance allowing bridging to occur. The revised mechanism predicts growth rates in good accord with
those measured experimentally and serves to support the methyl radical in a
growth role.
1.7 Applications of Synthetic Diamond
Synthetic
diamond produced by HPHT and CVD methods has proven useful in a number of
applications which, in general, utilise its extreme properties. While the use of diamond has replaced other
materials. The industrial use of
natural diamond is restricted by the relative scarcity and cost of obtaining
large pieces of diamond necessary for some applications. Even diamond grit used in the cutting tool
industry is produced by the HPHT process, as this is economically more viable
than the use of natural diamond.
Although both natural and HPHT diamond have found uses in a number of
applications these are limited, since the material is only available as single
crystal and the crystal cost increases exponentially with size. The use of CVD diamond deposition allows the
production of continuous diamond films that may be grown onto surfaces other
than diamond. This technology also
allows significantly more flexibility, as deposition from the gas-phase can
occur onto a number of different shaped and contoured surfaces.
1.7.1 Cutting and Grinding Tools
Diamond being
the hardest known material is the obvious choice for the cutting tool
industry. Natural, and more recently
HPHT, diamond has been used on coated cutting tools for a number of years. However single crystal diamond, while being
wear-resistant, is also prone to cleave easily. The use of polycrystalline CVD diamond as a coating on cutting
tools would overcome this problem. CVD
diamond coated cutting tools have however only recently become industrial
products due to problems associated with the adhesion of the coating[47]. Polycrystalline diamond (PCD), (a term used
in the cutting industry, not to be confused with the descriptive term used to
specify films polycrystalline in nature) commonly used in cutting inserts, is
formed via a multi-stage process. HPHT
diamond particles are compacted with cobalt to form a sintered disc which,
after treatment in a high-temperature high-pressure press, is then cut and
machined into inserts ready to be soldered onto cutting tools. PCD, although more robust than traditional
tungsten carbide, is not as wear resistant as tool inserts formed from CVD
diamond as table 1.2 illustrates.
|
Tool Type |
Lifetime /
minutes |
|
Tungsten Carbide |
<1 |
|
25 mm PCD on Tungsten Carbide |
40 |
|
30 mm CVD film on Tungsten Carbide |
60 |
|
0.5 mm CVD film on Tungsten Carbide |
70 |
Table 1.2 Relative tool lifetimes adapted from reference [48]. Refer to reference for information regarding test conditions
Diamond coated tool bits have proven to be versatile being used to machine stone, timber, reinforced plastics, aluminium and other non-ferrous metals. Diamond cannot be used to machine ferrous metals, nickel-based or titanium-based alloys because of its chemical reactivity with these materials at the high contact pressures and temperatures generated during machining.
1.7.2 Optical windows
Diamond
windows, having a broadband optical transparency, have recently become an
industrial possibility due to improvements in CVD technology that allow large
diameter optical-grade diamond windows to be produced. The broadband transparency, extreme thermal
properties and hardness of diamond lend itself to being used in a number of
high power laser or detector window applications.
Diamond’s
resistance to thermal shock and its transparency in the infrared region has
encouraged CVD diamond production for use as a window material in high power IR
lasers. Since diamond has a high
thermal conductivity its use in high power lasers prevents local heating of the
window, which in turn could lead to failure of the material. In order to prevent local heating of the
window the number of faults must be minimised during growth. Another use of diamond windows being
proposed is as an IR airborne sensor detector window for use on missiles. Since this may require use in hostile
environments (e.g. impact of hail, sandstorms, etc.) involving high wear,
diamond is now being considered as a substitute for more conventional IR window
materials such as zinc selenide[49].
Diamond
windows, being transparent to X-rays, are also finding applications as X-ray
windows in detectors and tubes. Diamond
may also be used in future lithography techniques for similar reasons[50].
1.7.3 Thermal Management
As the trend
for next generation high-power electronic and optoelectronic devices seems to
be towards smaller more compact units one major problem that needs to be overcome
is the efficient removal of large quantities of generated heat. By utilising diamonds unequalled thermal
conductivity, freestanding thick CVD films could be used as a medium to
‘spread’ the heat from the device to the cooling system. Diamond has another advantage over other
high thermal conductivity materials in that, being a non-metal, diamond is an
electrical insulator with obvious advantages in high-density electronic
packaging. CVD diamond films can now be
produced having a thermal conductivity in excess of 20 W cm-1 K-1
with dimensions suitable for use as a heat spreader in high-power electronics.
Recent
expansion in the telecommunications industry has involved the use of high-power
laser diodes, which produce a large quantity of heat. Failure of each unit by localised overheating would result in
huge financial losses. Diamond CVD heat
spreaders have been used as their relatively high cost is more than compensated
by the reduction in losses from unit failure.
1.7.4 Surface Acoustic Wave (SAW) devices
Surface acoustic wave filters are commonly used devices
present in televisions, video recorders, mobile telephones, etc. Introduced electric signal is converted to
sound, which is then reconverted into electric signal. This acts as a filtering system, as higher
frequency signal requires a higher frequency sound velocity. Diamond
has become of interest as a high velocity substrate material due to its elastic
constant which is the highest known of any material, with a SAW velocity of
more than 10000 m/s. This compares very
favorably with analogous ZnO/Sapphire devices (5500 m/s). Although diamond is not a piezoelectric
material, its high acoustic propagation makes it a desirable substrate for SAW
devices when coupled with piezoelectric thin films such as ZnO. Recent research at Sumitomo Electric[51]
has produced a Lithium-Niobate (LN) piezoelectric film on a diamond
substrate. It is suggested that the LN/diamond device will have a higher
electro-mechanical coupling coefficient than previous devices with high
acoustic velocity. Figure 1.9 shows a
schematic of a ZnO/diamond SAW filter.
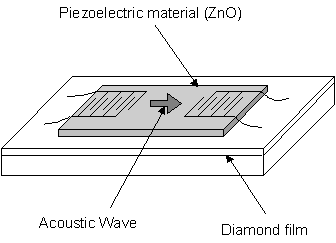
Figure 1.9
Schematic of a Surface Acoustic Wave device adapted from reference [52].
While conventional materials are limited by a frequency of 2.5 GHz, it is widely thought that diamond SAW devices will be used in the next generation of high-speed communications.
1.7.5 Detector devices
Conventional
photodetectors are generally sensitive to both visible and ultraviolet (UV)
light. This is usually as a result of the
band gap of the detector material.
Diamond, having a large band gap (5.49 eV), could effectively be used to
detect deep UV light while being blind to visible light. Being an extremely robust material, diamond
would be an excellent candidate for use in hostile environments, such as in
environmental monitoring or military applications.
While silicon
is commonly used as the detector material in particle accelerators, the next
generation of accelerators will need to be installed with more robust detectors. Interest has been shown in diamond due to
its resilience to damage[53]. There is also a potential for diamond alpha-
and neutron radiation detectors to become widely used in the nuclear
industry. It is likely that CVD diamond
devices will become available with high sensitivity and long lifetimes.
1.7.6 CVD diamond sensors and electronic devices
The many
extreme properties of diamond make it an excellent candidate material for
micromechanical devices, e.g. pressure and temperature sensors. Being extremely robust, and having an
unparalleled thermal conductivity, diamond seems the obvious choice over more
conventional wide band-gap semiconductors such as SiC and III-nitrides. Diamond, being an electrical insulator, may
be doped to form a p-type semiconductor.
This is commonly carried out by incorporation of boron, integrated into
the diamond lattice during the growth process.
Boron doped films have an acceptor level 0.368 eV above the valence
band. Problems with forming
controllable and stable n-doped diamond films by using the substitutional
dopants nitrogen or phosphorus have so far prevented the creation of diamond
p-n junctions. However, for most sensor
applications, one type of doping is sufficient.
The
resistivity of doped diamond decreases with increasing temperature; thus it may
be used as the active material in temperature sensors. Given its thermal conductivity, diamond may
also be deposited onto thermocouples providing a robust protective coating for
use in harsh environments without the introduction of significant measurement
errors.
The use of
doped diamond films has also been proposed as the active component in pressure
sensors. Thin silicon diaphragms, onto
which the diamond is deposited, flex according to the pressure exerted. The resistivity of the doped diamond film
will change as a function of applied stress, a consequence of the piezoelectric
effect. The resistance of the film may
then be converted into a pressure reading.
Semiconductor
manufacturers currently use electrolyte-based detection systems in order to
sense the accidental release of toxic doping gases such as PH3 and
AsH3. However, it is thought
that maintenance costs could be reduced with the introduction of solid-state
sensors. In order to be introduced the
active sensor material must be extremely sensitive while being selective
against other airborne impurities.
Recent preliminary reports on doped diamond-based devices have shown
very high degrees of sensitivity and species selectivity with good detection
reproducibility[54].
1.7.7 Diamond cold cathode emission devices
Cold electron emission is observed from metals when a
sufficiently high electric field is applied and electrons can overcome the
energy barrier to emission from the material.
Wide band-gap materials such as diamond have essentially no energy
barrier to electron emission; such materials are referred to having a negative
electron affinity (NEA). In NEA materials, once electrons are excited into the
conduction band, they are spontaneously emitted to the vacuum, as the vacuum
state is more stable.
Reports of
high quantum efficiency photoelectron emission from the (111) surface of
natural diamond suggest that diamond-based emission devices could operate at
very low power[55]. Research into emission devices using CVD
produced films has thus been encouraged.
The development of devices using this phenomenon is ongoing and may lead
to the production of flat-panel displays, vacuum microelectronics and microwave
amplifiers. The possible design of such
a device is shown schematically in figure 1.10.
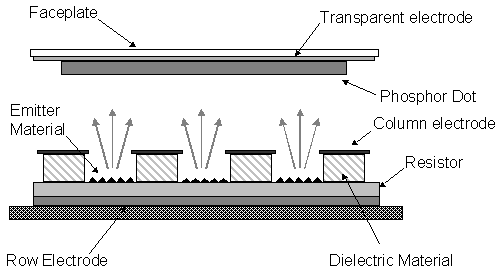
Figure 1.10 Schematic of a diamond–based triode structure cold cathode emission device.
While the
possibility of producing such a device seems likely, development has been
eclipsed by recent interest shown in similar devices using carbon nanotubes
(CNT) as the emitting material[56].
1.7.8 Other applications
A number of
niche applications have recently been suggested that utilise diamond’s extreme
properties.
The company
GFD specialises in the production of medical scalpels coated with diamond[57],
the scalpels are used in ophthalmic operations requiring extremely sharp
high-precision instruments. Diamond is
deposited via CVD methods onto a sharpened silicon blade thereby ensuring the
instrument remains sharp throughout its lifetime.
Due to
diamond’s high wear resistance its use has been suggested as a coating in high
wear environments such as the pumping components used in the recovery of crude
oil[58]. The pumping of large quantities of crude
oil, commonly containing large amounts of abrasive sand, has a detrimental
effect on pump components hence requiring frequent replacement. It is envisaged that diamond CVD coating of
components would prolong the lifetime of the apparatus thereby reducing costly
repair times.
One of the
first applications envisaged using CVD diamond was in the formation of speaker
diaphragms[59]. Speaker diaphragms require a stiff structure
and are formed from materials with high sound velocities. Natural diamond, with a sound velocity of
18.2 km s-1, compares favourably with competitive materials
(Beryllium: 12.6 km s-1, Aluminium: 5.1 km s-1) together
with an unsurpassed stiffness.
Manufactured diamond diaphragms, produced via hot filament CVD methods
onto a silicon substrate with subsequent etching, have exhibited sound
velocities of 16.5 km s-1 operating at a maximum frequency of 80,000
Hz, the highest generated by any dynamic speaker.
While previous
estimates of the impact of synthetic CVD diamond may have proven to be
over-enthusiastic[60] the last
ten years have shown a steady increase in the demand for CVD diamond as more
applications are found and the production costs decrease[61]. It seems inevitable that the use of CVD
diamond will continue to grow; particularly in the electronics industry.
1.8 CVD synthesis methods
A number of different diamond CVD methods have been developed over the last twenty years, but for a number of reasons, none of these techniques have totally replaced the more traditional hot filament system. Microwave and plasma jet CVD systems offer the advantage of increased growth rates as compared with hot filament systems, but the versatility, ease of production scale up and economic considerations associated with hot filament CVD reactors encourages their use in the majority of industrial production.
1.8.1 Hot filament CVD system
One of the
simplest, cheapest and to some extents most versatile methods of diamond CVD is
by a Hot Filament (HF) technique.
The major
features in a HF system are a resistively heated wire, allowing thermal
dissociation of H2, and a heated substrate. Figure 1.11 outlines a typical HF system.
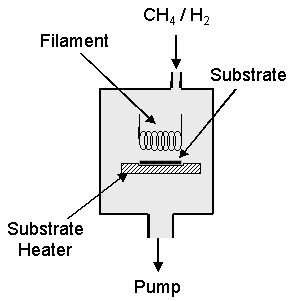
Figure 1.11 Schematic of a typical Hot filament system based on the NIRIM design
Originally
devised by the NIRIM group, and later developed by Matsumoto et al.[62],
the HF system used industrially today has changed very little. Typically a gas mixture of approximately 1%
CH4 in hydrogen is passed over a heated coiled filament maintained
at a temperature between 1800 and 2400°C. The filament
material maybe Tungsten, Tantalum, Rhenium or Molybdenum, the surface of which
forms a metal-carbide layer in the presence of the hydrocarbon-hydrogen
gas-phase environment. Thermal
dissociation of molecular hydrogen occurs at the filament surface to form
atomic hydrogen, which instigates the gas-phase chemistry. Atomic hydrogen, as mentioned earlier in
this chapter, is the main initiator of the hydrocarbon chemistry.
Diamond growth
occurs on the substrate surface, which is maintained at an optimum process
temperature by a substrate heater.
Growth rates of approximately 1 mm hr-1 are commonly achieved from a CH4/H2
input gas mixture using a typical HF system.
Although species transport typically occurs via diffusion from the
filament, growth rates of 5 mm hr-1
have been reported using forced convection[63].
HF CVD methods
suffer from problems associated with filament material being incorporated into
the grown film, the problem being more pronounced with high hydrocarbon gas
mixtures. Studies have shown that,
while the increase in hydrocarbon content promotes the growth rate, it also
causes increased deterioration of the filament surface. This will, in effect, prevent the growth of
high purity doped diamond films for electronic uses, hence the use of other
activation techniques (i.e. microwave enhanced CVD).
Due to HF
being the most available diamond CVD system, a relatively large number of
studies have been undertaken. These
studies have included investigations into the optimum process conditions
necessary for high growth and quality deposition. Zhou et al.[64]
has summarised a number of important growth factors.
- Increases in the nucleation density may be achieved by
pre-treating the substrate. This
leads to the rapid formation of a film due to the improved initial incorporation
of growth species.
- While the growth rate may increase with increasing
methane fraction in the gas feed (0.1-2%), the non-diamond content of the
film also increases.
- Maximum growth rate appears to occur at a chamber
pressure of ~10kPa and a substrate temperature of ~850°C.
- The growth rate appears to increase with increasing
filament temperature and decreasing filament-substrate separation. This may be attributed to increased
atomic hydrogen concentrations at the substrate surface.
Gas-phase studies
have attempted to answer a number of questions about the growth
environment. Laser-based diagnostic
techniques have been used to study the atomic hydrogen concentrations in a HF
reactor. Celii and Butler[65]
used Resonance-Enhanced Multiphoton Ionisation (REMPI) techniques (See Appendix
1) to study the production and destruction of atomic hydrogen in the gas-phase.
Probing 8 mm
from a tungsten filament at 2500 °C, they observed the atomic hydrogen number density to
decrease by an order of magnitude as the methane concentration increased from
one to three percent. Using
Laser-Induced Fluorescence (LIF), described in Appendix 2, Schäfer et al.[66]
reported a decrease of 30% in atomic hydrogen concentration when the methane
concentration increases to 5%. Atomic
hydrogen loss upon addition of hydrocarbon was also shown via Intercavity laser
absorption spectroscopy[67],
whereby, at low methane concentrations, small increments in the hydrocarbon
input resulted in a sharp decrease of the atomic hydrogen number density. Using Molecular-Beam Mass Spectrometry
(MBMS) sampling via a hole in the substrate surface, Hsu[68]
showed the concentration of atomic hydrogen to decrease by an order of
magnitude when the methane concentration in H2 increased from 0.4%
to 7.2%.
These results
all go some way to underlining the importance of atomic hydrogen in the
gas-phase chemistry. Atomic hydrogen
loss is attributed, to varying extents, to both H atom abstraction reactions,
outlined earlier in figure 1.4, and heterogeneous recombination.
Heterogeneous recombination of atomic hydrogen takes place via the third-body reaction 1.22.
2H +
M ®
H2 + M Reaction
1.22
Being a
third-body reaction the rate of recombination will be subject to the gas-phase
composition and the gas pressure.
Early growth
studies showed that diamond films may be grown at comparable qualities and
growth rates irrespective of the hydrocarbon feed gas[69]. This discovery led to studies aimed at
probing the gas-phase environment to ascertain the species present and to
provide evidence of the reactions occurring[70].
Studies of the
hydrocarbon gas-phase chemistry have focused on suggestions that the methyl
radical, CH3, or acetylene, C2H2, may be
viable growth species[71]. Celii et
al.[72]
used infrared diode laser absorption spectroscopy to detect both CH3
and C2H2 in a HF reactor. Vibrational spectra of C2H2, C2H4
and CH3 were measured in a HF activated 0.5% CH4 in H2
gas mixture at 25 Torr. Acetylene,
being the most stable hydrocarbon at lower reactor temperatures (i.e. far from
the filament), was shown to be the most abundant gas-phase species, while
concentrations of other stable hydrocarbons, such as C2H6,
C3H4 and C3H8, were below the
detection limit.
Using the
highly sensitive laser-based absorption technique Cavity Ring Down spectroscopy
(CRDS), described in Appendix 3, Zare et al.[73]
studied the absolute number density of CH3 radicals in the gas-phase
environment. Their results indicated a
peak in CH3 number density ~4 mm from the filament surface. The significance and validity of this result
is discussed further in Chapter 3, together with comparisons of REMPI
measurements obtained at Bristol.
These studies
have been backed up by theoretical simulation studies of the gas-phase
environment and the growing surface[74]. Without doubt, the HF CVD system is the most
studied of the diamond deposition techniques.
The need for a greater understanding of the gas-phase environment,
gas-surface interactions and the growth scheme remains in order to increase
understanding of the process and optimise deposition process conditions.
It is the
ability to scale-up the HF process that encourages its use in the diamond tool
coating industry. The need to coat
three dimensional tool edges and inserts requires a dynamic system such as
provided by HF CVD.
1.8.2 Microwave-plasma assisted CVD system
In a microwave-plasma assisted CVD (MPACVD) system, as the name suggests, microwave radiation (typically 2.45 GHz) is coupled into the gas mixture thereby forming and sustaining a plasma. Electron bombardment is a significant contributor to ionisation and decomposition of the gas. Efficient coupling of energy into vibrational levels allows gas temperatures of 2000-3000 K to be attained, prompting thermal dissociation of molecular hydrogen. As in HF CVD, atomic hydrogen initiates the necessary gas-phase chemistry. Figure 1.12 schematically outlines the main parts of a MPACVD system.
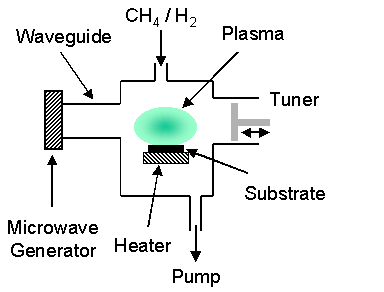
Figure 1.12 Schematic outlining the major components of a typical MPACVD system
While the gas
composition, pressure, and obtained growth rates are similar to that of a HF
system, microwave systems have the advantage of being able to produce large
diameter films. Since no electrodes or
filaments are used to decompose the gas mixture, the films grown are not
contaminated by metal impurities. This
has led to studies into the growth of MPACVD doped diamond films to exploit
their electronic properties[75].
The substrate
is usually positioned so as to make contact with the visible plasma
‘ball’. Diffusion is thought to be the
major transport mechanism by which active species reach the substrate. Bias-enhanced nucleation (BEN), whereby a
potential is applied to the substrate in order to enhance the nucleation stage,
is commonly used in MPACVD systems[76]. The BEN effect occurs due to the presence of
charged species produced in the plasma.
Since MPACVD
systems are not reliant on filaments or electrodes (which may deteriorate in
oxidising gas mixtures) to activate the gas-phase chemistry, a wider range of
input gases have been studied from the viewpoint of diamond deposition[77]. Of these, studies of oxygen addition in
particular have led to an increase in knowledge of the gas-phase chemistry
involved, as summarised in chapter 1.5.4.
1.8.3 Oxyacetylene Torch CVD
One way of
producing a sufficient number of active species for diamond deposition is via
the highly exothermic reactions that occur in flames. The Oxyacetylene torch CVD method, shown schematically in figure
1.13, burns a mixture of C2H2 and O2 to produce
a high enthalpy flame.
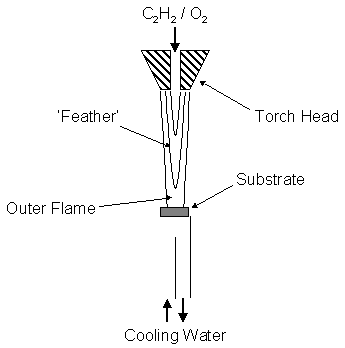
Figure 1.13 Schematic of a typical oxyacetylene diamond CVD system
The high
velocity flame impinges onto a water-cooled substrate and has been shown to
deposit high quality diamond[78]. The acetylene is oxidised to produce carbon
monoxide and atomic hydrogen in a highly exothermic reaction (DHreac ~ -448 kJ mol-1),
resulting in flame temperatures in excess of 3000 K. Carbon monoxide is stable at such high temperatures and any
unreacted acetylene may be converted into hydrocarbon radicals via reaction
with atomic hydrogen. For this reason,
diamond deposition is typically carried out with an acetylene flow marginally
greater than that of oxygen. This in
effect produces an optimum hydrocarbon radical concentration within a region of
the flame known as the ‘acetylene feather’.
Diamond deposition is carried out onto a substrate positioned within
this region.
Growth studies
have demonstrated the deposition of polycrystalline diamond at growth rates
approaching 200 mm hr-1. Films deposited by this technique suffer
from non-uniform radial growth, reflecting the radial variation in flame
composition and substrate temperature.
Attempts to overcome this problem via nozzle modifications and turbulent–flame
techniques have so far shown only minimal improvements[79]. The introduction of flat-flame burners has
been shown to enable uniform deposition over areas up to 13 cm2 and
such burners have started to replace the more typical welding torch design[80].
Extensive
studies using Laser Induced Fluorescence (LIF) have been carried out focusing
on radial distributions of C2, CH, OH and CN within the flame[81]. While the gas-phase chemistry is very
different from that in typical methane-hydrogen CVD environments, the actual
growth mechanism is thought to be the same.
Since this
technique is relatively inexpensive to set-up and, in terms of deposition,
extremely flexible, its use has been postulated for thick film coating of
objects that have niches or curved surfaces[82]. Unlike all other deposition techniques,
oxyacetylene torch CVD may be carried out at atmospheric pressure without the
use of vacuum equipment. This has
obvious advantages over HF and microwave CVD coating methods.
1.8.4 Plasma-jet CVD system
The term
Plasma-jet refers to a medium to high pressure CVD system whereby convection is
the main transport mechanism, energy may be provided by a number of means. Radio-frequency (RF) inductively coupled
plasma jets and microwave plasma assisted plasma jets have both been shown[83]
to deposit high quality diamond films.
In general, direct-current (DC) plasma jets are most commonly used.
Figure 1.14
shows a schematic illustration of a typical DC plasma jet system used for
diamond deposition. DC plasma jets
(also known as DC-arcjets) operate by passing an electric arc discharge through
a gas flow, which via ohmic heating increases the gas enthalpy and kinetic
energy, thereby producing a plasma jet (plume). For diamond deposition, the plasma jet system typically operates
on an Ar/H2/CH4 gas mixture, with the hydrocarbon usually
being added downstream of the torch head to prevent the deposition of amorphous
carbon on the exit nozzle. The plasma
jet expands into a reaction chamber maintained at a medium to high pressure and
impinges onto a water-cooled substrate typically maintained between 1000–1500
K.
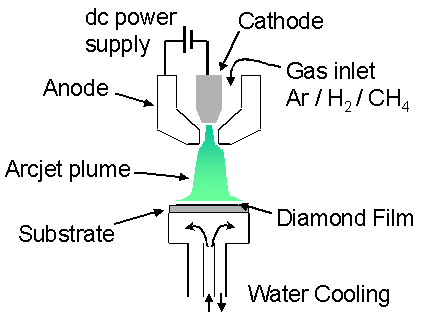
Figure 1.14 Schematic outlining the major components of a diamond depositing DC-arcjet CVD system. Adapted from reference [84].
The expansion
of the plasma jet is thought to be supersonic in some systems[85]
with the plume velocity being dependant on both the design of the torch head
and the pressure difference between the torch head and the main chamber into
which the plume expands. As the plume
expands into the main chamber the pressure difference promotes the gas dynamic
conversion of thermal energy into kinetic energy to form the plume. The high convection velocity of species
within such a system is thought to account for the high growth rates obtained.
Since
convection dominates the species transport the residence time within the
reactor is strictly defined. This
provides a significant conceptual advantage when modelling plasma jet systems,
as compared with other CVD methods in which diffusion is the primary species
transport mechanism and therefore the residence time is less clearly defined.
The high
average gas temperature within the plume exceeds that obtained via thermal
activation methods as used in HF CVD.
As a result of the high gas temperatures the decomposition of molecular
hydrogen to form atomic hydrogen, a species fundamental to diamond deposition,
is more complete.
Since the
first demonstration of DC-arcjet diamond deposition by Kurihara et al.[86]
published in 1988, a number of groups have endeavoured to optimise process
conditions, determine growth characteristics and lower film production
costs. In 1990 Ohtake et al. demonstrated what remains the
highest reported linear growth rate from a gas precursor using any CVD
technique by using a DC plasma jet system[87]. The study achieved a growth rate of over 900
mm hr-1
with a carbon conversion efficiency of approximately 8%. However, by using a hydrocarbon liquid
precursor injected through the substrate even higher growth rates of ~1mm hr-1
have been demonstrated[88].
A range of
other techniques have been shown to further improve both the rate and quality
of film deposition. For example, by
using a secondary discharge, positioned above the substrate, an increase in
both film quality and growth rate is achieved[89]. It is thought that the secondary discharge
maintains an increased atomic hydrogen concentration above the substrate, hence
counteracting its recombination in the boundary-layer.
Growth rates
from the reported studies are extremely varied; this may be clarified when the
large differences in input power are taken into consideration. The growth rate has been shown to scale with
the input power used[90]. Surprisingly, the content of non-diamond (or
the defect density) was also found to increase as a function of input
power. The study concluded that the
increase in input power increased the concentration of atomic hydrogen, which
is consequently consumed in activating hydrocarbon radicals, leading to an
enhanced C:H ratio (in excess of the optimal value), thereby causing an
increase both in the growth rate and the defect incorporation in the growing
film.
The number of
gas-phase studies of plasma jets during diamond deposition is considerably less
than for the more common CVD systems, but significant progress has nonetheless
been made using a number of diagnostic techniques.
Kawarada et al.[91]
used optical emission spectroscopy (OES), described in appendix 4, to ascertain
some of the species present within the plume.
OES is restricted to detection of emitting species only and is subject
to collisional quenching, for these reasons it is difficult to determine
reliable species concentrations solely from emission data. Another OES study by Loh and Cappelli[92]
concluded that the high diamond growth rate is due to high gas flow rates and
rapid quenching of the plasma at the substrate. Rapid quenching of the high temperature plasma (>5000 K) was
thought to produce super-equilibrium levels of hydrocarbon radicals.
Using both LIF
and OES to ascertain plasma temperatures,
Gicquel et al.[93] were able to refine a gas-phase model, thus providing one of
the earliest and most fundamental studies of DC plasma jet diamond
deposition. The group incorporated a
stagnation boundary layer at the substrate into their hydrodynamic model and,
in conjunction with diagnostic measurements, found the plasma temperature to
fall linearly from a maximum of 5000 K to 2100 K at the stagnation boundary in
front of the substrate and to 1200 K at the substrate surface.
Plasma
diagnostics found the electron and gas temperatures to be almost identical,
leading to the conclusion that the plasma was essentially thermalised. By utilising the hydrodynamic model, an
atomic H mole fraction of 0.460 was calculated at the boundary layer, indicating
that approximately 25% of the feedstock H2 dissociates. Once produced, atomic hydrogen and carbon
both persist in the plasma since consumption of these species involves a
three-body reaction (from inspection of data in reference 25). Given the high atomic H concentrations in
the plasma, it is unlikely that methyl radicals persist long enough in the
gas-phase to propagate to the substrate at high enough concentrations to
account for observed growth rates of over
100 mm hr-1. In their study the group therefore suggested
that atomic carbon might play a role in diamond growth, with the majority of
the total carbon balance being converted into polyatomic hydrocarbon species in
the cooler regions peripheral to the plasma.
Further
evidence of the plasma being close to thermalised in a low-pressure (20 Torr)
regime was revealed by findings that the electron temperature was only a factor
of two greater than the gas temperature[94]. Larger discrepancies in the electron and gas
temperatures are commonly seen at higher pressures. One other aspect of this study also identified a plateau in the
gas temperature with increasing input power, an observation attributed to the
absorption of energy by the reaction, H2 ® 2H.
Modelling the
SRI reactor using an experimentally determined temperature profile, together with
gas-phase chemistry, stagnation flow hydrodynamics and diamond surface
chemistry, Goodwin was able to calculate the atomic hydrogen concentration as a
function of position, including close to the substrate surface[95]. At a distance of 0.2 mm from the substrate
surface the atomic hydrogen mole fraction was calculated to be approximately
0.2, corresponding to about 11% dissociation of the molecular hydrogen
feed. In contrast to earlier studies,
Goodwin was able to show that calculated CH3 concentrations (~2.0´10-4
mole fraction) were sufficiently high to account for the observed high growth
rates in DC plasma jet CVD. Atomic
carbon concentrations were shown to be about a factor of 10 higher than that of
methyl radicals in the gas-phase close to the substrate.
While these
reports are informative, and go some way to providing an initial insight into
the gas-phase chemistry involved in the diamond depositing DC plasma jet
system, it is worth noting that the early reactor designs are fundamentally
different to the modern reactor. The
realisation that remote addition of the hydrocarbon downstream from the torch
head both improves the deposition rate and prevents the build up of amorphous
carbon within the torch head, has promoted the use of remote hydrocarbon addition. Improvements in modelling carried out by
Dandy and Coltrin[96] took into
account gas-surface chemistry, and the effect of a swirling gas flow upon
stagnation flow hydrodynamics. One of
the conclusions from this influential study was the identification of two
diamond growth regimes, which it was suggested, are dependant on the degree of
H2 dissociation in the gas phase.
For schemes where H2 dissociation is low (~5%), CH3
is thought to be the dominant growth species, whereas in the event of high H2
dissociation (up to 95%), it was postulated that atomic carbon is the dominant
growth species, with the growth rate increasing with the extent of H2
dissociation.
While any
reported model will be tailored to a specific reactor and deposition conditions
a number of general trends have emerged.
In a combined diagnostic and modelling study Reeve et al.[97]
identified a number of similarities in the trends of gas-phase species
concentrations in previously published work.
By using the CHEMKIN gas-phase chemistry modelling package24
the group calculated the CH3 radical concentrations near the nozzle
of the torch head. They found CH3
to be the dominant hydrocarbon within 5 cm of the nozzle implying that this
region corresponded to the low H2 dissociation regime. Further from the nozzle exit, further H atom
abstraction occurs, leading to formation of other hydrocarbon species. As the Reeve et al. group typically
deposit diamond onto a substrate positioned, at most, 5 cm from the torch head
nozzle, their study goes some way to underline the role of methyl radicals in
diamond growth in this plasma jet environment.
Reeve et
al. have also postulated that systems using remote hydrocarbon addition,
being subject to inefficient mixing of the hydrocarbon with the plasma, may in
effect lower the residence time of active species in the hot plume. Typically CH4 is used as the
input hydrocarbon which, via H atom abstraction, forms CH3. The CH3 radical may in turn
suffer H atom abstraction (as shown in figure 1.4). It was suggested that the residence time might be sufficiently
reduced by using a remote hydrocarbon addition system and so maximise CH3
production. Loh and Cappelli[98]
also attempted to study the effect of reducing the species residence time by
increasing the gas flow rates (in a study involving CH4 addition
through the torch head).
Thus it is
clear that, in order to optimise diamond deposition from CH3, a low
residence time is required to minimise further abstraction reactions. This would be easier to prove if it were
possible to quench the hydrocarbon chemistry by reducing the available atomic
hydrogen. However, H atoms at the
substrate surface are vital for high quality diamond growth. Therefore, an optimum H2
dissociation level must exist within this regime for a given species residence
time, so as to achieve the necessary compromise between maximising CH3
concentrations, while maintaining sufficient H atom flux at the substrate.
Mass
spectrometry has been used to monitor the exhaust gases from a diamond depositing
DC plasma jet reactor, the major stable reaction products being CH4,
C2H2 and C2H6 [99]. Little information of the gas-phase
chemistry involved in diamond deposition may be gained via mass spectrometric
analysis of the exhaust gases, the hot plume preventing any in-situ sampling.
Electrostatic
probe analysis has been used to ascertain the electron temperature and
densities within a diamond-depositing plume96. Reeve and Weimer used a floating double
probe technique, yielding respectively, electron temperatures and electron
densities of 2.3 eV and 7´1011
cm-3 near the nozzle exit and 0.4 eV and 1´109
cm-3 downstream. This study
shows the plasma to be weakly ionised concluding that reactions involving
electrons must only play a minor role in diamond deposition. The DC plasma jet in this study was
generated by a 2.8 kW discharge with expansion into a chamber maintained at 60
Torr. This study represents a low-power
system and while both the electron temperature and density in the plume will
increase with increasing input power, it is still thought that reactions
involving electrons are not essential to diamond deposition.
By using a
Langmuir probe, Stalder and Jefferies[100]
measured the electron temperature (Te) and density as a
function of process gas composition in a
1-2 kW
discharge plume expanding into a chamber maintained at 25 Torr. Collisionless thin-sheath probe theory was
used to interpret the I-V curves
obtained. In the high velocity plasma
jet the mean free path will be significantly greater than the Debye length,
validating the use of collisionless theory.
This study showed very little variation in Te as a function of distance from the nozzle exit. The group also measured the ion density and Te to be respectively 6x1011
cm-3 and 1.1 eV at a distance of 13 mm from the nozzle exit
operating on a diamond depositing gas mixture (49% H2 / 51% Ar /
0.2% CH4).
Furthermore,
the group found that the electronic properties of the plasma deviated from that
predicted by the Saha equation, with the electron density being more than six
orders of magnitude greater than that calculated. The Saha equation, described in detail in Appendix 5, relates the
electron density to the ionisation potential and electron temperature, assuming
Boltzmann equilibrium applies to both the neutral and ionised species of the
plasma. The deviation in the value from
that predicted using the Saha equation signifies non-equilibrium between the
electron-ion density and the gas and electron temperatures. In order to produce such a non-equilibrium
situation it was concluded that, while species ionisation is primarily carried
out in the activating arc, the resulting free electrons are transported
downstream via convection.
Brinkman et
al.103 have drawn comparisons between species emission and ion
densities within the plume. By studying
the emission intensity and ion density as a function of distance from the
nozzle exit, it was shown that the CH and H (Balmer-g) emission
falls by a factor of 25 to 30 from 13 to 42 mm downstream of the nozzle exit,
while the ion density drops by a factor of 10.
The emission from the C2(d) state measured over the same range showed a smaller decrease,
approximately a factor of three. It was
concluded that while emission from H and CH is by means of electron impact
excitation, C2 emission must arise by another means. It has been postulated therefore that
chemiluminescent reactions account for most of the C2 emission.
Storm and
Cappelli[101] have
carried out similar studies finding electron densities of approximately 1013
cm-3 in a 1.4 kW hydrogen arcjet.
While this level of ionisation is about ten times higher than that
measured by Brinkman et al., it is thought to be consistent with
a decrease in ion density with axial distance.
In the study
of diamond depositing DC plasma jets, OES is the most commonly used diagnostic
tool. This non-invasive spectroscopic
technique has been used to analyse the composition of the high luminescent
plume by a large number of groups [102],[103]. OES relies on the collection of spontaneous
emission from a species in an excited state.
The experimental set-up of each OES study is fundamentally the
same. Emission is collected along an
axis at right angles to the plasma flow, defined into a column, and conveyed by
a fibre optic into a spectrometer.
Wavelength dispersed emission is collected via a charge-coupled device
(CCD) array or a photomultiplier tube positioned at the exit of the
spectrometer. Within the visible
wavelength range
350-700 nm,
emissions from the species C2, CH, H, C, Ar and, where applicable,
CN and CO are detectable. This has led
to studies measuring the emission intensity of these species as a function of
process conditions, in order to ascertain rotational temperatures and species
trends. However, quantitative species
analysis is complicated by collisional quenching and species self-absorption
within the plume.
OES studies
performed by Reeve, Weimer and Cerio[104]
concentrated on the rotational and vibrational excitation temperatures of C2
and CH. Spatially resolved emission
spectra were taken to ascertain species temperatures in a plane between the
nozzle exit and the substrate. Plasma
excitation temperatures were determined by fitting calculated optical emission
spectra associated with the d3Pg ® a3Pu transitions of
C2 and the A2D®X2P transitions
of CH to measured spectra. Only a
scaling factor, the vibrational temperature (Tvib) and the
rotational temperature (Trot) were used as fitting parameters
in a least-squares fit of the observed and calculated spectra. To derive a meaningful temperature from an
emission spectrum the emitting species must be thermalised.
As deviation
from thermodynamic equilibrium is common in reacting plasmas, small differences
between electronic, vibrational and rotational temperatures and the gas
temperature are expected. Any
differences in these temperatures from the optical emission are directly linked
to the excitation and de-excitation processes that produce non-equilibrated
excited state populations. The
competing de-excitation processes include spontaneous emission, rotational and
vibrational energy transfer and collisional quenching. Results of the fitting procedure showed a
good agreement between the C2 and CH observed and calculated
spectra.
Determinations
of the Trot and Tvib showed that while the
plasma is not thermalised i.e. Trot ¹Tvib, a general temperature trend existed
as a function of distance from the substrate.
Reeve et al. thus concluded that excited state C2 and
CH are likely to be strongly quenched by H2. They also concluded that the excitation
mechanism for C2 and CH are very different, resulting in widely
different rotational and vibrational excitation temperatures.
It is thought
that in the bulk plasma, chemiluminescent reactions are responsible for
producing CH emission, while rotational and vibrational temperatures,
determined from optical emission spectra, indicated that C2
excitation occurs via electron impact.
Close to the substrate, it was thought that thermal excitation is most
likely to be the major excitation mechanism for both CH and C2, as
the plasma closely resembles a hot neutral gas.
In a study
carried out by Cubertafon et al.[105]
emission spectroscopy was used to produce rotational temperature maps of the
plume. Spatially resolved rotational
temperatures of the CH (B2S®X2P) band
revealed a relative plateau in the temperature profile from the nozzle exit,
where
Trot ~2600 ± 200 K, to a
point approximately 5 mm from the substrate surface where the temperature drops
rapidly towards the substrate temperature (~1200 K). This trend has been substantiated by a pulsed laser Rayleigh
scattering study[106]. Emission from atomic carbon (3s:1P®2p2:1D
and 3s:1P®2p2:1S)
was shown to be stable throughout the distance from the nozzle exit to close to
the substrate surface where it decreased.
Cubertafon et al. hypothesized
that atomic carbon is therefore consumed at the growing surface as a precursor
to diamond growth. This finding is
consistent with a number of models which identify atomic carbon as a growth
species[107],[108].
Results of
measuring the electron density from the Stark broadening of the H Balmer lines
have also been reported. Due to the
large electron flux within the plasma jet, Stark broadening of emission lines
is observed and may be used to estimate the local electron density110. H Balmer-a emission was collected and fitted in
terms of a Voigt profile with a Gaussian contribution removed to allow for
instrument broadening. The resultant
broadening of the emission line was shown to be strongly dependent on the torch
anode-cathode separation. Electron
densities, obtained from the FWHM (Full-Width Half Maximum) of the Voigt
profiles, are estimated to be in the region of
1015-1016
cm-3 close to the nozzle exit of a torch operating between 3-5 kW on
an Ar/H2/CH4 gas mixture.
While no data
were published, it was stated that the electron density falls as a function of
distance from the nozzle exit. The
report notes, that while the line broadening (and hence electron density) is
influenced by the anode-cathode separation, the quality and growth rate of the
films are not. This provides further
evidence that the precise value of the electron density plays a minor role in
determining the gas-phase environment prior to diamond deposition.
Attempts have
been made to estimate the electron temperature from the relative emission
intensities of the atomic hydrogen Balmer series[109]. The method used by Luque et al. assumes the excited states of
atomic hydrogen have similar electron impact excitation cross-sections and collisional
quenching rates. The emission intensity
is then related to the electron temperature via equation 1.2,
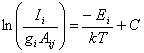 Equation 1.2
Equation 1.2
where Ii is the emission intensity from
level i, gi is the degeneracy of this level, A is the Einstein A coefficient for the i®j transition, Ei is the energy, k the Boltzmann constant, T the electron temperature and C is a constant. In previous studies[110]
it was found that the departure from thermodynamic equilibrium deems this
technique unreliable.
By using
Laser-Induced Fluorescence (LIF) techniques, discussed in Appendix 2, absolute
number densities for a series of species have been estimated[111],[112]. By probing the ground (or low-lying excited)
state, LIF has the advantage over OES of providing absolute species number
densities unaffected by deviations in thermal equilibrium. Luque et
al. have used LIF to determine spatial distributions of C2, C3
and CH. It was shown that C2
and CH both have a maximum radial number density in the centre of the plume, while
C3 is concentrated in a hollow cylinder about the jet axis. As previously shown by OES, CH number
density was found to maximise about 1 mm from the substrate.
Rotational and
vibrational temperatures of the detected species were also measured and compared
with a rotational temperature gained from NO, a non-reacting species. Differences of ± 200 K were
found in the free stream of the plume for CH rotational and vibrational
temperatures and the rotational temperature of NO. Within the axial centre of the plume a local gas temperature of
2300 ± 150 K was
obtained. Comparisons between optical
emission-based measurements and LIF measurements of temperature have been drawn[113].
OES
measurements from rotational and vibrational excited CH and C2
radicals yielded temperatures in the range 3000-7000 K, whereas LIF
measurements of the vibrational and rotational state populations in the
respective electronic ground states, gave temperatures in the 1200-2200 K
range. These discrepancies merely serve
to underline the problems associated with the non-equilibrium nature of the
plasma, and some of the difficulties in interpreting plasma properties from the
optical emission.
It is worth
noting that while a number of general trends exist in both the gas-phase and
surface chemistries, as well as the hydrodynamic properties within DC plasma
jets, each diagnostic measurement and model calculation will be specific to the
reactor used. Contributions to any
difference in observed parameters may come from torch head and reactor design,
together with deposition conditions such as input power, input gas mixture, etc.
DC plasma jets
have an advantage over other CVD techniques in that the deposition of diamond
can proceed simultaneously with the deposition of ceramics and metals. Kurihara et al.[114]
deposited a 40 mm thick
diamond film onto a tungsten-molybdenum substrate, by depositing a preliminary
layer of tungsten carbide followed by a diamond-tungsten carbide layer. Such graded material reduced the thermal
stress between the diamond film and the substrate, and increased the overall
film adhesion strength by an order of magnitude.
Commercially,
the use of a DC plasma jet system to deposit diamond has not proven to be as
successful as other techniques. The
initial cost of a DC plasma jet system is considerably higher than competitive
microwave and hot filament reactors.
Since plasma jets require the consumption of large quantities of both
power and process gases (argon and hydrogen) it is not thought currently to be
economically viable except for the production of small diameter thick
freestanding films. Recent studies by a
Chinese group have shown how gas recycling may lower the film production costs[115]. These studies demonstrated a recycling
efficiency of 85%, with a concomitant reduction in diamond production costs
from approximately $45 to $21 per carat.
Regardless of
the economic drawbacks of DC plasma jet deposition, the unsurpassed growth rate
and high quality deposition explains why plasma jets continue to attract interest
as a deposition technique.
References