Chapter 4 :
Experimental and Theoretical studies of a nitrogen
containing HF-CVD
gas-phase environment
The addition of small quantities of nitrogen to
typical hydrocarbon/H2 gas mixtures used during diamond CVD, in both
hot filament and microwave reactors, has been shown to lead to enhanced
deposition rates[1],[2],
nitrogen addition appears to affect the growth habit and lead to some modest
nitrogen incorporation in substitutional lattice sites[3].
Previous investigations at Bristol by May et al.[4]
focused on diamond CVD in a hot filament reactor using CH4/NH3/H2,
CH3NH2/H2 and HCN/H2 gas
mixtures. The aims of these studies
were to see whether use of alternative nitrogen containing precursors provided
a route to enhanced nitrogen incorporation in the grown diamond film, Further
aims were to unravel aspects of the chemistry prevailing in H/C/N containing
gas mixtures using in-situ molecular
beam mass spectrometry (MBMS), to sample the gas-phase composition in the
vicinity of the hot filament.
The study observed diamond deposition from CH4/NH3/H2
feedstock gas mixtures, provided the input gas ratio [CH4]:[NH3]
³ 1 was observed. The addition of NH3 was found to reduce deposition
rates relative to those found for a simple CH4/H2 gas
mixture. CH3NH2/H2
and HCN/H2 gas mixtures were also shown to yield CVD diamond,
with low efficiencies comparable to that found using 1:1 mixtures of CH4
and NH3 in H2.
Such findings are explicable given the MBMS measurements, which
showed substantial conversion of the input carbon to HCN (the thermodynamically
favoured product), at the relevant process temperatures[5]. Thus it was proposed that, in the vicinity
of the hot filament, HCN acts as a sink for carbon that might otherwise have
participated in diamond growth. The
findings detailed in this chapter therefore build on the conclusions drawn by
May et al.
This chapter describes a combination of experimental
and 3-D computer modelling in order to provide a more detailed interpretation
of the effects of controlled additions of nitrogen (in the form of N2
and NH3) to CH4/H2 process gas mixtures, in an
HF-CVD reactor. The experimental
component involves use of REMPI spectroscopy to provide spatially resolved in-situ measurements of H atom and CH3
radical number densities, and served to inspire subsequent Cavity Ring Down
Spectroscopy (CRDS) measurements of NH radical column densities with the same
gas mixtures, in the same HF-CVD reactor, by J.B. Wills, as a function of
process conditions (e.g. feed-stock
gas mixing ratio and the temperature, Tfil,
of the Ta filament). These REMPI
measurements are a natural extension of our investigations of diamond CVD when
using CH4/H2 and C2H2/H2
gas mixtures in the same HF-CVD reactor detailed in chapter 3. The results obtained are then compared with
the output of a 3-D computer model, specifically tailored to this HF-CVD
reactor.
The 3-D computer model has been described in detail
in chapter 3, and therefore only changes to the previous modelling procedure
will be discussed here. The gas phase
chemistry and thermochemical input is provided by the GRI-Mech 3.0 detailed
reaction mechanism for C/H/N/O gas mixtures (plus one additional reaction
allowing destruction of the species H2CN), but with all reaction
steps and species involving O atoms removed (shown in Appendix 6). Comparisons between experimental and theory
highlight the very different reactivities of N2 and NH3,
in the present HF activated CH4/H2 gas
mixtures. These comparisons also help
unravel details of the gas-phase chemistry prevailing when using H2/CH4/NH3
process gas mixtures, and reveal clear evidence for a reduced H atom production
rate, Q, (described in chapter 3) on
the surface of the HF, when NH3 is added to the process gas mixture.
The results of the CRDS study of NH[6]
are also shown in this chapter purely to emphasize the accuracy of the 3-D
model.
Details of the HFCVD reactor, and the REMPI detection
schemes used for spatially resolved measurements of H atom and CH3
radical number densities, have been presented in chapter 2
and are essentially unchanged, save for accommodating the introduction of N2
and NH3 into the reaction chamber.
The H2, CH4, NH3 and/or N2
feedstock gases are metered through separate mass flow controllers, pre-mixed
in a manifold, and enter the reactor through a port located above the cradle
assembly, so as to maintain an overall flow rate of 100 sccm and total
pressure of 20 Torr.
Addition of NH3 to a CH4/H2
gas mixture activated by a Ta hot filament was observed to cause a
reduction in the Tfil
value returned by the two-colour optical pyrometer. No such trend was observed upon addition of a corresponding flow
rate of N2. Fig. 4.1
illustrates these effects, via plots of Tfil
versus time after the introduction of various partial pressures of NH3 (and
N2) to an established 1% CH4 in H2 gas
mixture.
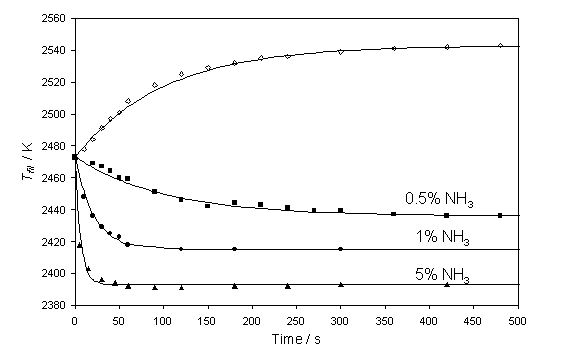
Figure 4.1
Variation in pyrometer measured Tfil as a function of
time after introducing 0.5% NH3 (¢), 1% NH3 (˜) and 5% NH3 (▲) to a 1% CH4
in H2 gas mixture. The solid
lines are best fits to these datasets in terms of equation 4.3 with t = 100 s (0.5% NH3), 21 s (1% NH3)
and 6 s (5% NH3). Also shown
is the time-dependent recovery of Tfil on the retardation of
a 2% NH3 flow (¯) together with a best fit with a time constant of t = 100 s.
In each case, the filament had been run for
> 6 hrs with a CH4/H2 gas mixture prior to
the NH3 (or N2) addition, and the power supplied to the
filament was held constant throughout.
Clearly, both the rate and the extent of the temperature drop, DTfil (defined as the difference between Tfil measured prior to any NH3
addition, at t = 0 s, and the asymptotic value found at t = ¥) increase with increasing NH3 fraction,
whereas addition of 1% N2 has minimal effect on the observed Tfil. Each of the Tfil versus t
trends measured for the various NH3 partial pressures can be
described by a function of the form:
Tfil(t) - Tfil(t = ¥)
= DTfil exp(-t/t) Equation
4.1
with a time constant, t, that decreases as the NH3 fraction in
the input gas feed is increased. Fig.
4.1 also illustrates how this temperature recovers with an apparently
invariable time constant when the NH3 flow is shut-off (¯). Prior to
any NH3 addition, we foresee the filament as being largely
'carburised', i.e. to be mostly tantalum carbide with a surface that is
partially covered by a coating of graphitic carbon. Given knowledge of the input power supplied to the HF (Pinput = 87 W (~12.4 V, 7.0 A) for Tfil(t = 0)
= 2473 K in figure 4.1) and estimates of the power expended on H2
dissociation (2 W) and conductive losses (10.7 W) from the 3-D
modelling, we can estimate the power radiated by the HF (~74 W). This, in turn, allows estimation of the mean
emissivity, e, of the hot filament via Planck's radiation law,
Prad
= e s S Tfil4, Equation
4.2
where s is the Stefan-Boltzmann constant and S is the calculated surface area of the
filament (0.63 cm2). The
emissivities derived, e ~ 0.56 and ~0.61, for the addition of respectively 0% and 1% NH3
fall midway between literature values for the emissivity of TaC (e ~ 0.3)[7]
and graphite (e ~ 0.9)[8]
at T ~ 2500 K. This implies that, under these conditions,
approximately half the filament surface has a graphitic coating.
The observed time dependence of Tfil upon addition of NH3 may be expressed in
terms of dynamic competition between the adsorption, desorption and/or etching
of the hot filament surface by nitrogen and carbon containing entities. In this scheme, two limiting mechanisms
merit consideration. The first assumes
that, both N and C containing species are present, and that the former are
adsorbed preferentially onto the filament surface. 'N-termination' on the filament is presumed to alter the energy
balance and increase the mean emissivity, leading to the observed reductions in
Tfil and in Q
(detailed in section 4.4).
Assuming this is so, the observed time dependence of Tfil (shown in equation 4.1)
can then be rationalised by considering a simplified adsorption-desorption / etching kinetic scheme, in which
accommodation of a N containing gas phase species on an available surface site,
S*, leads to N-termination, i.e. via
the dynamic equilibrium,
N
+ S* ¾ SN. Equation
4.3
The time dependence of, q, the N-terminated surface coverage, will then
follow,
q = ![]() (1 - exp[-(k5[N]
+ k-5)t] Equation
4.4
(1 - exp[-(k5[N]
+ k-5)t] Equation
4.4
where [N] is the number density of nitrogen containing
species near the filament surface, and k5
and k-5 are,
respectively, the forward and reverse reaction rate coefficients for the
generic gas-surface reaction described in equation 4.3. Fitting the experimental data for addition
of 1% and 5% NH3 to a 1% CH4 in H2 gas mixture
with
Tfil = 2473 K (at t = 0), shown in figure 4.1,
yields values of k-5 ~ 0.01
s-1 and
k5[N] =
0.38 s-1 and 0.157 s-1, respectively. The observed recovery of Tfil
when the NH3 flow is discontinued may also be calculated using equation
4.4.
It is also worth mentioning that similar decreases in
Tfil have been reported
previously in the case of carburised Ta filaments operating at high
temperatures (Tfil >
2800 K) and constant Pinput
when the hydrocarbon content in CH4/H2 gas mixtures is
suddenly reduced[9]. This
may seem counterintuitive at first, since a reduction in the number density of
gas-phase carbon would be expected to induce a concomitant reduction in the
extent of any graphitic overcoat on the filament surface, which should
therefore lead to a reduced emissivity and thus cause Tfil to rise.
This observation has been explained by realising that the graphitic
‘overcoat’ is considerably less efficient than a 'clean' TaC surface at
catalysing H2 dissociation; the observed drop in Tfil is thus attributed to
the increase in the fraction of Pinput
expended on surface catalysed H2 dissociation.
As shown previously, one effect of NH3
addition is to convert CH4 molecules into HCN in hotter regions of
the gas. HCN is a stable molecule in
this environment, and thus acts as a 'sink' for gas-phase carbon near the
filament surface. Therefore, a second
possible model for the time dependence of Tfil upon NH3
addition presumes that, NH3 addition has the effect of reducing the
number density of gas-phase carbon available for formation of any graphitic
layer on the filament surface. This
would have a similar effect to simply reducing the CH4 input flow
rate. This, in turn, might then be
expected to cause an increase in S*, capable of catalysing H2
dissociation and could thus also give rise to the observed time dependent
variations in Tfil. Given our earlier estimates of the fraction
of Pinput used in
dissociating H2 molecules on the filament surface, at the
comparatively low Tfil values used in this work, such an
explanation seems incapable of accounting for Tfil reductions of the magnitudes observed upon NH3
addition.
4.3 Gas-phase H atom and CH3
radical relative number densities observed with the addition of NH3
and N2
Relative number densities of H atoms and CH3
radicals were measured by 2+1 REMPI (in an identical manner to that used in
chapter 3), as a function both of added NH3 (or N2) and Tfil. Figures 4.2 and 4.3 show experimentally
measured [H] and [CH3] values, as a function of added N2
and NH3, for a 1% CH4 in H2 mixture (flow rate
= 100 sccm, total pressure = 20 Torr and Tfil
= 2573 K), measured at a filament distance d = 4 mm.
Clearly, given the above change in filament
characteristics, it was necessary to increase the power supplied to the HF
progressively as the NH3 input mole fraction increased in order to
maintain a constant Tfil.
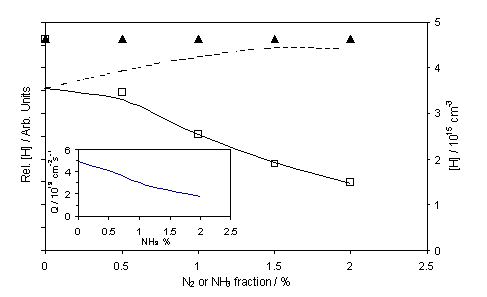
Figure 4.2
REMPI measured H atom relative number density, as a function of added NH3
(£) and N2 (p) to a 1% CH4 in H2 gas
mixture. The chamber was maintained at
20 Torr and the filament at Tfil = 2573 K. The dashed curve shows the calculated
variation in absolute [H] (right hand scale), assuming fixed values of
Q
(4.92 ´ 1019 cm-2s-1) for Tnf
= 2075 K using the previously established Tgas dependency on d. The solid curve however shows a fit to the
experimentally determined NH3 data assuming that Q declines
with added NH3 with a function shown in the figure inset. The theoretical and experimentally
determined data were vertically scaled so as to match for 1% NH3
addition.
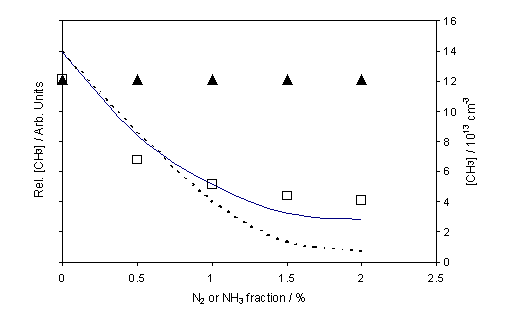
Figure 4.3
Equivalent plot showing REMPI measured CH3 relative number
density as a function of added NH3 (£) and N2 (p) to a 1% CH4 in H2 gas mixture. The reactor conditions are identical to that
stated for figure 4.2 with measurement also carried out at d = 4
mm. The dashed and solid curves
represent the calculated variation in absolute CH3 number density
given a fixed Q, and when assuming that Q varies as a function of
added NH3 as shown in the inset of figure 4.2.
Interpreting these, and subsequent, experimental data
builds on previous modelling of the gas phase chemistry occurring in activated
CH4/H2 and C2H2/H2 gas
mixtures in this same HF-CVD reactor (Chapter 3). The modelled reactor (with no substrate present) is represented
in Cartesian co-ordinates with the z-axis
parallel to the direction of feedstock gas flow (input ® output) and perpendicular to the filament, the
centre axis, of which, is along y. x
is orthogonal to both the filament axis and the direction of gas flow, the
point (0, 0, 0) defines the centre of the filament, and d = 0 corresponds to the point
(0, 0, 1.5 mm). The model
calculation extends to the volume enclosed within
x = ± 16 mm, y = ± 18 mm and z = ± 26 mm, with the gas temperature set to Tgas = 600 K at all
boundaries of the numerical grid. The
positions (20 mm, 0, 0) and (0, 20 mm, 0) represent the reactor walls
while (0, 0, -25
mm) and (0, 0, +25 mm) represent the gas outlet and inlet
positions.
The dashed lines in figures 4.2 and 4.3 show absolute
H atom and CH3 radical number densities predicted by 3-D
simulations, assuming, the experimental values for Tfil and the gas pressure, a net H atom production rate at the filament
(Q = 4.92 ´ 1019 cm-2 s-1), a
near filament gas temperature (Tnf
= 2075 K) and a d dependence for Tgas consistent with our
previous measurements. In both figures,
the measured [H] and [CH3] values appear insensitive to the addition
of N2. This result can be
readily understood by inspecting the calculated temperature (and thus position)
dependent interconversion rates and number densities for the various
participating species. In the case of a
1%CH4/1% N2/H2 gas mixture, N2 is
deduced to play the role of an essentially inert spectator, with a predicted
fractional dissociation [N]/[N2] < 10-7 even at Tgas ~ 2000 K (i.e. as attained in the immediate
vicinity of the HF). Further
simulations, in which this ratio was arbitrarily increased (such as could
conceivably apply in the event of catalysed N2 dissociation on the
filament surface or, in the case of microwave activation, as a result of
non-thermal plasma chemistry), indicate that the [N]/[N2] ratio
would have to be at least 100 times higher in order to cause a measurable
reduction in [H] or [CH3].
However, the addition of NH3 into the hot
filament activated 1% CH4 in H2 gas mixture, clearly
changes the gas-phase chemistry. The
calculated z dependent
interconversion rates and number densities for all major species present in a
hot filament activated
1% CH4 / 1% NH3 / H2
gas mixture, displayed in figure 4.4, show this nitrogen containing precursor
to be intimately involved in the high temperature gas-phase chemistry. Temperature and pressure dependent rate
constants, k(T,P), for the numerous
elementary steps underpinning the H/C/N gas phase chemistry are taken from the
GRI-Mech 3.0 reaction mechanism. The
addition of the highly exothermic reaction,
H
+ H2CN ®
H2 + HCN, Equation
4.5
which was originally excluded from the GRI-Mech
database due to its overall minor role in combustion analysis, curtailed the
calculated build-up of unstable H2CN in the cooler regions of the
reaction chamber.
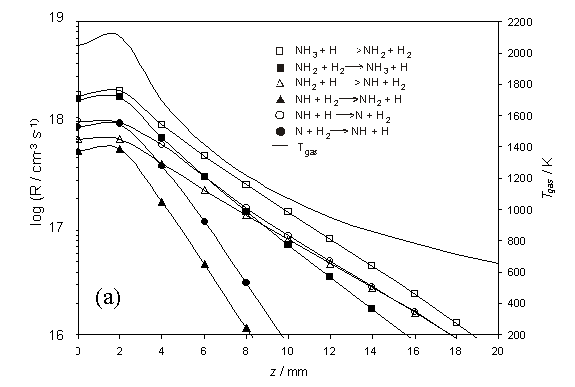
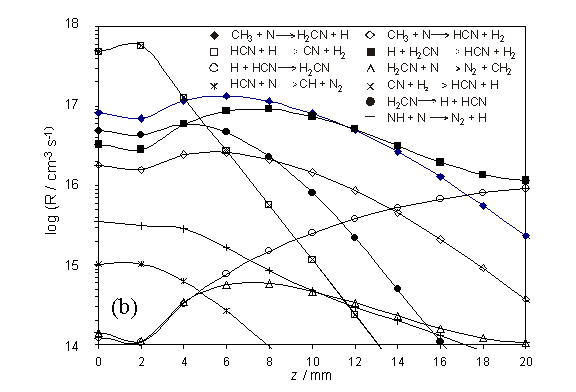
Figure 4.4
Plots showing the semi-logarithmic calculated rates, R (left-hand
scale), for (a) the series of hydrogen shift reactions, NHx + H ![]() NHx-1 + H2, and (b)
other selected reactions dealing with nitrogen containing species. Both plots are shown as a function of
distance z (with x = y = 0) for a chamber operating with
1% CH4 / 1% NH3 / H2 and Tfil
= 2573 K. The R values were
calculated assuming Q = 2.0 ´ 1019 cm-2 s-1, Tnf
= 2100 K and a z dependent temperature profile as shown in (a)
(right-hand scale).
NHx-1 + H2, and (b)
other selected reactions dealing with nitrogen containing species. Both plots are shown as a function of
distance z (with x = y = 0) for a chamber operating with
1% CH4 / 1% NH3 / H2 and Tfil
= 2573 K. The R values were
calculated assuming Q = 2.0 ´ 1019 cm-2 s-1, Tnf
= 2100 K and a z dependent temperature profile as shown in (a)
(right-hand scale).
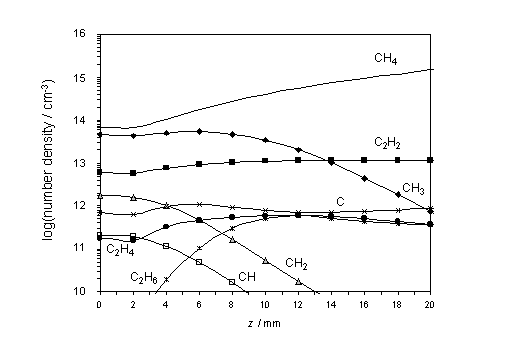
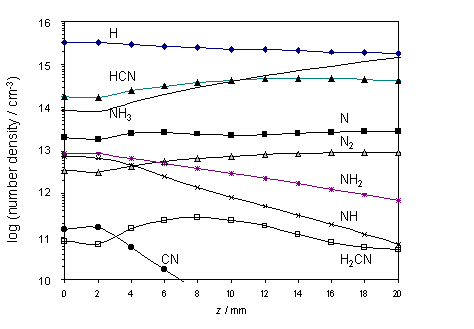
The calculated species
number densities, clearly show HCN acting as a carbon ‘sink’ in both the hot
and cool regions of the chamber. This
is shown in figure 4.5 together with other relevant species.
![]()
![]()
The experimental results, shown in figures 4.2 and 4.3,
and the calculated rate constants and species number densities, shown in figure
4.4, are for a constant Tfil,
gas pressure and flow rate. As the
dashed lines in these figures show, no single value of Q is capable of
fitting all data recorded as a function of added NH3. To match the experimental observations it is
necessary to assume Q to be NH3 dependent, i.e. to
assume that N-termination of the HF surface causes not just an increase in
emissivity but also a reduction in the mean H atom production rate, Q.
This much improved agreement is illustrated by the
solid lines in figures 4.2 and 4.3, which demonstrates that it is possible to
obtain a reasonable fit with the experimentally measured [H] and [CH3]
versus Tfil dependencies,
if Q for the filament, maintained at
a constant Tfil, declines with the percentage of added
NH3. The deduced functional
form of Q(NH3; Tfil = 2600 K) is shown in
the inset to figure 4.6. Such a
dependence may be considered plausible given the previous conclusions that NH3
addition leads to modification ('N-termination') of the filament surface and of
its emissivity.
4.4 Gas-phase H atom and CH3
radical number densities in a 1%CH4/1%NH3/H2
gas mixture as a function of Tfil
Figure 4.6 compares H atom relative number densities
measured 4 mm from the filament surface as a function of Tfil for a 1%CH4/H2 input gas
mixture, with and without 1% added NH3, while figure 4.7 shows the
corresponding plots for the measured CH3 radical relative number
densities.
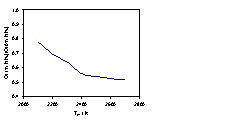
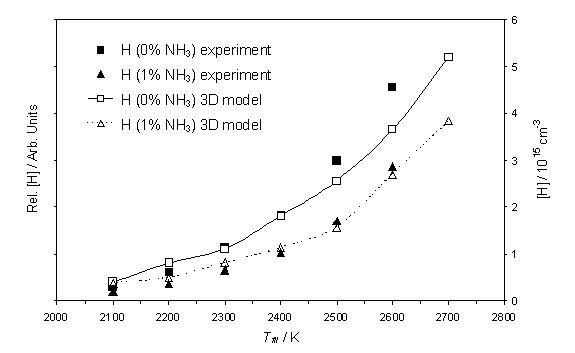
Figure 4.6
Calculated [H] and measured relative H atom number density as a function
of Tfil. Both the
calculation and the measurements were carried out using a 1% CH4 / H2
gas mixture, with and without the addition of 1% NH3, at 20
Torr. REMPI measurements were carried
out at d = 4 mm.
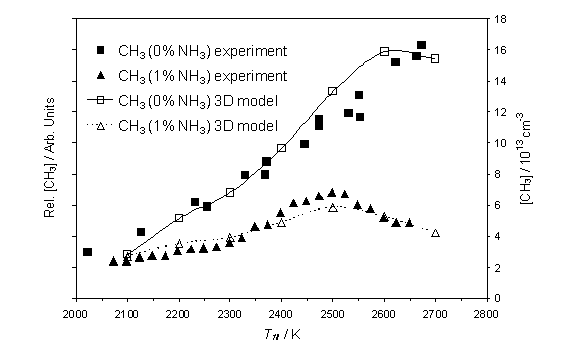
Figure 4.7
Calculated [CH3] and measured relative CH3 number
density as a function of Tfil. Both the calculation and the measurements were carried out using
the same conditions as described in figure 4.6.
As shown in previous experiments in Chapter 3 (Figure
3.6), the H atom number density increases almost exponentially as a function of
increasing Tfil. The
observation that the decrease in the H atom number density, with the addition
of NH3, continues throughout the filament temperature range is also
shown. As in figure 4.2, model
calculations based purely on gas-phase chemistry, together with the assumption
of a constant Q, suggest that [H] should increase upon addition of NH3. In order to obtain convergence between
experimental observation and the model calculations it is necessary to assume
that the NH3 induced modification of Q is itself a function
of the filament temperature. As figure
4.6 shows, a reasonable match between experiment and theory can be obtained if
the ratio Q(1%NH3)/Q(0%NH3) is assumed to vary
with Tfil. Figure 4.7 shows, in accord with previous
findings, that [CH3] rises smoothly with increasing Tfil for the standard 1% CH4/H2
mixture, but that addition of 1% NH3 causes a marked decrease in [CH3],
most notably at high Tfil. Both trends are reproduced well by the 3D
model calculations but, again, better agreement with the trends measured in the
presence of 1%NH3 can be obtained by assuming that Q(1%NH3)/Q(0%NH3) is Tfil
dependent.
The value of Q may be seen as a function of
factors attributed to Tfil and the NH3
fraction. Let us consider both factors
in terms of H production on the filament surface. Q may be related to Tfil via the
equation,
Q(Tfil)
~ exp(-DHdiss/RTfil) Equation 4.8
4.5 NH radical number densities
and spatial profiles in 1% CH4 / x% NH3 / H2 gas
mixtures.
Analysis of the modelling results of the z dependent species concentrations and
elementary reaction rates, detailed in figure 4.5, indicate that the number
density of NH radicals in the vicinity of the hot filament (>1012
cm-3), should be sufficient to permit their detection by a sensitive
absorption method like CRDS. Unlike
REMPI, such measurements offer a route to absolute column densities, and thus
provide a further test of the absolute values of Q, and their dependence on Tfil
and [NH3], returned by the gas phase chemistry modelling. Such measurements have been undertaken by
J.B. Wills6. As with the
REMPI measurements, number densities were determined as a function of radial
distance from the lower edge of the coiled filament, and as a function of input
NH3 mole fraction. Figure 4.8
compares the measured and predicted filament distance, d, dependence of
NH radicals in a hot filament activated 1% CH4 / 5% NH3 / H2 gas
mixtures with Tfil = 2473 K, monitored via the R1(2)
and R1(3) transitions of the NH(A-X) origin band.
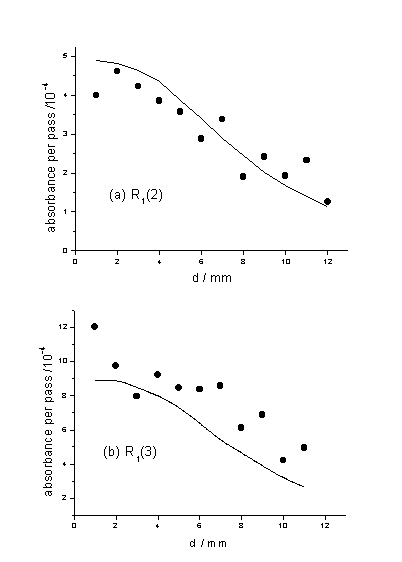
Figure 4.8
Measured (˜) and calculated(¾) d dependence of the NH absorbance. The measured data were obtained by
monitoring (a) the R1(2) and (b) the R1(3) transitions of
the NH(A-X) origin band.
Naturally the NH absorbance, and thus [NH], is found
to scale essentially linearly with the input NH3 mole fraction. The
level of agreement (to within ~20% in the implied absolute number densities)
provides strong support for the validity of the foregoing modelling of the gas
phase chemistry of H/C/N containing mixtures in a HFCVD reactor, and the
spatially resolved absolute number densities returned by the simulations for
all major species.
4.6 Conclusions
The results presented here, and elsewhere, provide a
reasonably self-consistent picture of the gas phase transformations occurring
in a HF CVD reactor operating at typical modest filament temperatures when
trace amounts of NH3 are added to a hydrocarbon/H2 gas
mixture. Input CH4 molecules
pass through a multi-step sequence of reactions en route to stable products C2H2
and HCN, thereby depleting the number densities of CHx species
both near the HF and in the surrounding volume, extending out to distances
where the substrate would normally be positioned in a diamond growth
experiment. HCN also acts as a sink for
input NH3 molecules. The
studies detailed here show that NH3 additions to an activated CH4/H2
gas mixture lead to reductions in the CHx number density in the
vicinity of the HF in two ways.
Firstly, addition of NH3 introduces additional, purely
gas-phase, CHx loss processes.
Secondly, NH3 additions are deduced to cause modifications of
the HF surface, increasing its emissivity and thus lowering Tfil (for a given input
power) and reducing its efficiency for catalysing H2 dissociation
even if Tfil is maintained
constant.
Additional confirmation
of the REMPI measurements and the modelling results was provided by CRDS
analysis of NH radicals, which afforded knowledge of the radical radial number
density dependence as a function of the distance from the filament.
It is worth noting that
the modelling study predicts CN radical concentrations at typical filament –
substrate distances to be < 109 cm-3. If, as thought, nitrogen incorporation into
a diamond film is carried out via CN addition into a vacant surface site, this
may provide an explanation for low nitrogen doping efficiencies.
References