

1.1 Diamond
To
the public at large, diamond has long been valued as a precious stone,
conjuring up images of many faceted brilliant gem stones, wealth and special
occasions. However, to the scientist,
this sp3-bonded allotrope
of carbon (Figure 1.1) is of particular interest due to its extraordinary
physical properties. These include the
following:1.1
n exceptional
hardness
n wide spectral
range transparency
n chemical
inertness
n high thermal
conductivity
n highest atomic
number density
n highest elastic
modulus
n very low
coefficient of expansion
n low coefficient
of friction, comparable to Teflon
n biological
compatibility
n when doped
becomes semiconducting
n Good electrical
insulator
n Very resistant to
chemical corrosion
n exhibits low or
‘negative’ electron affinity
Given
these many notable properties it is not surprising to learn that diamond has
already found uses in many variant applications such as heat sinks, abrasives1.2,
and wear-resistant coatings for cutting tools.1.3 Given its other unique properties it is
possible to envisage that diamond has momentous potential as an engineering
material in many other applications.
However, progress in implementing many such ideas has been restricted by
the scarcity of naturally occurring diamond.
It has therefore long been desired by scientists to devise methods to
synthesise diamond in the laboratory.
Figure
1.1. The Diamond Lattice Figure
1.2. The Graphite Lattice.
At ambient temperatures and
pressures, graphite, the sp2-bonded
allotrope of carbon (Figure 1.2), is the stable allotrope of carbon. The standard enthalpies of formation of
diamond and graphite differ only by 2.9 kJ mol-1,1.4
but a large activation barrier rules out simple thermal activation as a means
of driving the graphite®diamond
interconversion. Since diamond is the
densest allotrope of carbon, at high pressure, diamond must be the stable form
of solid carbon. This is the scientific
basis for the high pressure high temperature (HPHT) techniques by which the so
called ‘industrial diamond’ has already been synthesised commercially for over
30 years.1.1,1.2,1.5 The
advent of Chemical Vapour Deposition (CVD) of diamond has dramatically
increased the possibility of exploitation of its other extraordinary properties
and potential wide-ranging industrial applications. This technique uses, as process gases, a standard hydrocarbon gas
typically methane) in an excess of hydrogen.1.3,1.6,1.7 The resultant diamond produced by this
technique can display properties comparable to those of natural diamond.
1.2
Historical overview of the CVD diamond process
The
synthesis of diamond has been pursued ever since the French chemist Antoine
Laurent Lavoisier discovered that diamond was a form of crystalline carbon.1.8 The first report describing successful
diamond synthesis using HPHT methods was published in 1955,1.9 in
which diamond is crystallised from metal solvated carbon at pressures and
temperatures in the region of 55,000 atmospheres and ~1800°C.
Experiments with low
pressure synthesis had been ongoing in both the United States and USSR during
the same period. The first patents
emerged as early as 1962 by Eversole of the Union Carbide Corporation1.10
who reported diamond synthesis under reduced pressure by the CVD
technique. His process involved the
growth of diamond on diamond substrates with the use of carbon-containing gases
at pressures of less than 1 atmosphere and temperatures of around 800-1000°C.1.11 Eversole had actually succeeded in late
1952, even before results of HPHT techniques were published. This technique, however, was not very
effective since large amounts of graphite were co-deposited with diamond, and
growth rates were extremely low.
Eversole’s work was followed up by Angus and co-workers who provided
evidence of the use of atomic hydrogen as an etchant for graphite.1.12 In the mid-1970’s, the Soviet group led by
Spitzyn, Bouilov and Derjaguin showed that growth on non-diamond substrates,
such as copper, silicon and tungsten, was possible.1.13 This is a significant achievement as growth
had only previously been possible on diamond substrates. The Japanese built on both these
developments to devise a ‘hot filament’ CVD technique1.14 in which
atomic hydrogen was incorporated in to the growth process to give good quality
films on a number of substrates with significantly better deposition rates,
making diamond film growth commercially viable for the first time. Widespread interest developed from here, and
at present a variety of CVD deposition techniques have been established.
1.3 The
diamond CVD technique
Chemical
vapour deposition involves a gas-phase chemical reaction taking place above a
solid surface, which results in deposition onto that surface. All the CVD techniques employed to deposit
diamond films require a means of activating gas-phase carbon-containing
precursors. This involves mainly
thermal (e.g. hot filament) or plasma (microwave or R.F.) activation, or use of
a combustion flame (oxyacetylene torch).1.7 Figure 1.3 shows two of the most common
deposition techniques and gives some indication of typical operating
conditions.
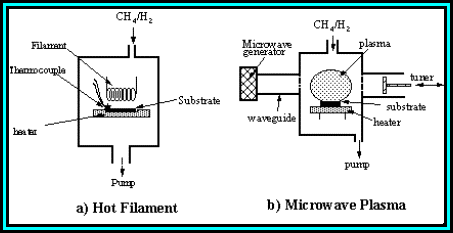
Figure 1.3. Examples of two most common types of low
pressure CVD techniques to deposit diamond. (a) Hot filament reactor, and (b)
Microwave plasma enhanced reactor.
Typically any
hydrogen/hydrocarbon source gas mixture may be used, subject to the C/H ratio
being less than ~0.03. Common to each
deposition technique and source gas mixture employed in the synthesis of CVD
diamond is the requirement of high gas temperatures which generates atomic
hydrogen and produces reactive carbon species.
Furthermore the substrate temperature must be maintained in the range
1000-1400K. The resulting films are
polycrystalline, with a morphology that is sensitive to the precise growth
conditions (See Section 1.4). The
deposition rate differs from one CVD deposition technique to another, and it is
generally observed that higher deposition rates can be achieved at a cost, and
that is the ‘quality’ of the films. The
quality of a CVD diamond film is normally judged by a number of measures such
as the ratio of sp3 (diamond) to sp2-bonded (graphite)
carbon in the sample, the composition (e.g.
C-C versus C-H bond content) and the crystallinity. In general, combustion methods deposit diamond at high rates
(typically 100 mm/hr), but are
restricted to small substrate areas, and with poor process control leading to
poor quality films. On the other hand,
hot filament and plasma methods produce high quality films but at much slower
rates (0.1-10 mm/hr). One of the main aims in current CVD research
is to increase the growth rates to economically viable rates (hundreds of mm/hr) without compromising
film quality.
1.4 The CVD
diamond film
The
surface morphology resulting from diamond CVD depends critically on
C/H ratio in the source gas mixture, as well as the substrate
temperature. At low substrate
temperatures and low CH4 concentration, diamond films are produced
with predominantly triangular {111} facets with many twin grain boundaries (See
Figure 1.4).

Figure 1.4. Surface
morphology of a diamond film grown on Si.
The film is polycrystalline, with twinning and many crystal defects
present.
{100} facets, appearing as
both square and rectangular forms, become more evident as the concentration of
CH4 in the source gas mixture, and/or the substrate temperature, is
increased. Cross-sections of these
microcrystalline films shows the growth to be mainly columnar in nature (See
Figure 1.5)

Figure 1.5. Cross section through a 6.7 mm-thick diamond film on Si,
showing the columnar characteristics of the growth up from the surface.
At
even higher CH4 concentrations the crystalline morphology disappears
altogether; instead a film composed of an aggregate of nanocrystals and
disordered graphite is formed, as shown in Figure 1.6.

Figure 1.6. Nanocrystalline film, exhibiting
‘ballas’-type morphology, typical of diamond grown under high (>2%) CH4
concentrations. This film is much
smoother than the microcrystalline film.
1.5 The
choice of substrates suitable for growing CVD diamonds
The
most commonly used substrates used to deposit CVD diamonds are Si wafers, but other
substrate materials may also be used.
What these substrate materials share in common is the need to withstand
the temperature window (1000-1400°C) required for
diamond growth. This precludes the use
of existing CVD techniques to diamond-coat materials such as plastics or
low-melting metals like alumimium. It
is also useful that the substrate be capable of producing a carbide. Initial stages of CVD diamond growth on
non-diamond substrates would involve the formation of a carbide interfacial
layer upon which the diamond then grows.
It is difficult to grow on materials with which carbon is ‘too
reactive’, i.e. those with which
carbon exhibit a high mutual solubility (e.g.
transition metals such as iron, cobalt, etc.). Substrates like Si, Mo and W are by far the
most popular because these materials form carbides, but only as a localised
interfacial layer due to their modest mutual solubility with carbon under
typical CVD process conditions. The
carbide layer can be envisaged as the ‘adhesive’ which promotes growth of the
CVD diamond, and aids its adhesion by (partial) relief of stresses at the
interface. The popularity of Si as the
substrate material is also due to its comparatively low thermal expansion coefficient,
which again serves to minimise the build up of stress in the CVD diamond films.
1.6
Mechanism of CVD growth
A
diamond CVD process is schematically summarised in Figure 1.7.1.16 Gaseous precursors, typically 1% CH4
in H2 at ~20 Torr, flow onto the reactor and gas-phase
reactions are initiated by the hot filament or plasma. The reactants, products, and reactive
species are transported throughout the reactor by diffusion and
convection. On the surface, adsorption,
diffusion, reaction and desorption of various species occur giving rise to the
nucleation of diamond particles, suppression of graphitic carbon, and
ultimately the growth of a continuous diamond film.
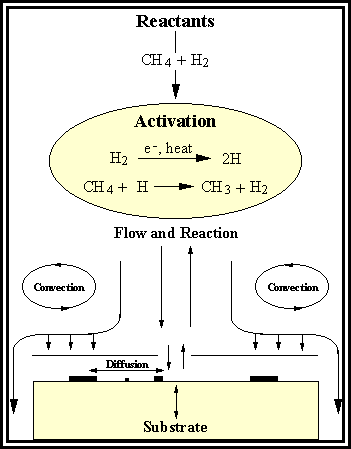
Figure 1.7. A Schematic diagram showing the principle
elements in the complex diamond CVD process: flow of reactants into the
reactor, activation of the reactants by thermal and plasma processes, reaction
and transport of the species to the growing surface, and surface chemical
processes depositing diamond and other forms of carbon (Adapted from Reference
1.16).
1.7 Role of
atomic hydrogen in the CVD process
As
previously mentioned, graphite is thermodynamically the more stable form of
solid carbon at ambient pressures and temperatures. Atomic hydrogen is believed to change the relative free energies
of small graphitic and diamond nuclei, thus providing a lower energy pathway
for the conversion of graphite to diamond.
Therefore diamond is the predominant product of the CVD process.1.12,1.17 H atoms are produced either thermally (at
the filament) or via electron bombardment; once formed they are believed to
play a number of crucial roles in the CVD process:
n They undergo H
abstraction reactions with stable gas-phase hydrocarbon molecules, leading to
the formation of highly reactive carbon-containing radical species. Unlike the stable hydrocarbon molecules,
these active radical species can diffuse to the substrate surface and
react. As a result, the all important
C-C bonds are formed, leading to the propagation of the diamond lattice.
n Atomic hydrogen
etches both diamond and graphite but, under typical CVD conditions, the rate of
diamond growth exceeds its etch rate whilst the reverse is true for all other
forms of carbon (e.g. graphite). This is believed to be the basis for the
preferential deposition of diamond over graphite.
n H-atoms terminate
the ‘dangling carbon bonds on the growing diamond surface and prevent them form
cross-linking, thereby reconstructing to a graphite-like surface.
1.8
Gas-phase chemistry involved in the CVD process
The initiation of the gaseous chemistry is dominated by dissociation of molecular hydrogen into atomic hydrogen, which is achieved either by thermal dissociation on a hot filament or by electron impact dissociation in a plasma. The subsequent gaseous chemistry is driven by reactions of atomic hydrogen with hydrocarbon species and a series of complex reactions among the hydrocarbon species. Figure 1.8 shows a schematic of this complicated ‘chemical soup’ of the major elements, reactive atoms and radicals.
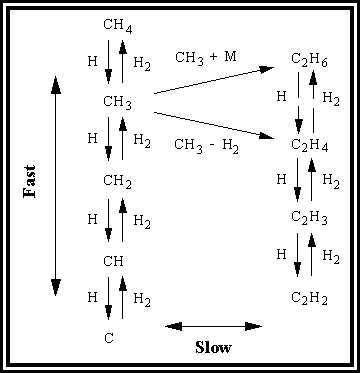
Figure 1.8. A schematic diagram showing the major
elements involved in the complex mix of reactions. The reactions involve rapid hydrogen transfer reactions amongst
the C1 and C2 species, and to a lesser extent the
bimolecular hydrocarbon reactions forming C2 and higher species.1.16
Since
typical process pressures are below 1 atm H atom recombination is slow,
and a super-equilibrium concentration of hydrogen atoms is present.1.18 Furthermore, since the major species in the
gas phase are H and H2, and the total hydrocarbon concentration is
about 1%, the hydrogen transfer reaction rates are generally much greater than
those describing the bimolecular hydrocarbon reaction rates.1.19-1.21
1.9 Characterisation
of the CVD process
There
is considerable interest in understanding the mechanism involved in the CVD of
diamond films, which could lead to greater control of a potentially beneficial
process. However, the precise mechanism
leading ultimately to the growth of a CVD diamond film is still not very well
understood. Several groups have
attempted to determine the gas-phase species involved in the process, but no
definitive answers have been reached.
Several techniques, both optical and physical diagnostic methods have
been employed, such as optical spectroscopy,1.20,1.22-1.24 resonance-enhanced
multiphoton ionisation (REMPI),1.25 gas chromatography,1.26,1.27
cavity ring down spectroscopy (CRDS)1.29,1.30 and mass spectrometry.1.31-1.34 It is mass spectrometry which is of most
interest to the present work. Using a
mass spectrometer it is possible to detect a number of species simultaneously,
and accurately quantify their composition (See Chapter 3). Harris and his co-workers1.34
utilised a quartz probe to sample gaseous products from a hot filament CVD
diamond chamber during deposition, which were then analysed using a mass
spectrometer. However, a direct
measurement of radical species such as methyl (CH3) or ethenyl (C2H)
radicals was not possible because of recombination of these species in the
probe used to sample the gases. An
attempt was made to estimate the concentrations of both radicals by calculating
ions from measurements of the concentrations of the likely recombination
products, namely C2H2 and C2H6
respectively. The main drawback to this
method is relying on the fact that they assume that there are no alternative
recombination reactions that could occur with other radical species. A means of detecting radical species
directly from the growth region is therefore of fundamental importance in
gaining a true identification of the gas-phase species responsible for the
propagation of diamond during the CVD process.
The solution is the molecular beam mass spectrometry (MBMS) system
developed by Hsu et al.1.35 which has enabled the detection of
stable as well as free radical species.
A three stage differential pumping system was utilised to cause a large
pressure difference to exist between the CVD chamber (20 Torr) and the mass
spectrometer (10-8 Torr).
Gas from the reaction chamber is extracted through a small orifice
(typically 100 mm in diameter) to
the high vacuum region of the mass spectrometer. The pressure difference induces the formation of a supersonic
molecular beam that effectively freezes out reactions, even between reactive
species such as free radicals. This
beam has an unobstructed path towards the ionisation chamber of the mass
spectrometer where, ultimately, the observable species are analysed.
1.10 Summary
of the work performed on hot filament CVD systems.
Much
research relevant to the present work has been performed by Harris, who
utilised mass spectroscopic technique.
The overall conclusion is that CH3 and C2H2
are the most likely candidates responsible for diamond deposition, because they
are the two main observable product species present under optimum growth
conditions. This results accord well
with chemical kinetics modelling1.22 which predicts comparatively
large concentrations of both species.
Subsequent work1.34 by the same authors involved measuring
the mole fractions of the stable species CH4 and C2H2
in the diamond growth environment. As
previously mentioned, recombination at the probe used to sample the gas
precludes the detection of CH3, but an indirect method was employed
to determine the methyl concentration by measuring the C2H6
and C2H4 species concentrations. However, these species were detected in low
quantities, presumably because of their transient nature at high H atom
concentrations, [H]. At optimum growth
conditions, C2H2 is the dominant C2 species
in the gas phase. Since the formation
of [C2H2] depends on [CH3], the detection of
gas-phase acetylene is now generally taken as an indicator of high steady state
[CH3] and hence of diamond growth.
Further
research1.31 was performed to examine the importance of methyl
radicals and acetylene. Aided with a
chemical kinetics model1.22 it was proposed that CH3
and/or CH4 is an effective diamond growth precursor. Furthermore they observed that the diamond
films deposited using CH4/H2 source gas mixtures were of
better quality and higher growth rates than when C2H2 was
the precursor gas. In addition, it was
believed that C2H2 was possibly responsible for the
deposition of non-diamond carbon phases.
This observation, coupled to the fact that the effective reaction
probability for diamond growth (defined as the number of carbon atoms deposited
as diamond divided by the number of collisions between a growth species and the
diamond surface) due to CH3 was greater than that due to C2H2,
led the authors to believe that CH3 is the major growth precursor in
hot filament CVD process. In a later
report1.36 it was found that the rate of diamond deposition was
directly proportional to the calculated mole fraction of CH3 present
in the reactor, but no such relationship was observed for the calculated C2H2
mole fraction. This supports the view
that methyl radicals are important intermediates in the CVD diamond growth
mechanism.
The
development of the MBMS system by Hsu1.37 has enabled the detection
of radical species as well as stable hydrocarbon species in the CVD growth
process. The apparatus allowed
measurements of H, CH3, CH4 and C2H2
as a function of CH4 concentration in the source gas mixture. Results show that hydrocarbon species other
than those above were present in trace amounts regardless of input CH4
concentration, and therefore are unlikely to be involved in the growth
mechanism. At low [CH4] in
the feed gas the dominant product is that of acetylene although at higher [CH4]
methane becomes the main hydrocarbon species.
A rise in the CH3 concentration was also observed with [CH4],
though to a lesser extent at high methane input. The measured H atom mole fraction, by contrast, drops rapidly
with increasing CH4 feed, presumably due to filament poisoning
effects (See Section 8.7, Chapter 8).
As a result, reactions which require the presence of [H] in the gas
phase, such as the formation of CH3 and H abstraction reactions of
the C2 hydrocarbon species (ultimately forming C2H2) will
occur much less readily, leading to a decrease on the absolute concentration of
both CH3 and C2H2. This is accompanied by an increase in the graphitic content in
the deposited diamond films with methane feed, which confirms the importance of
the role of H atoms in etching graphite and other non-diamond carbon phases
(See Section 1.7).
1.11
References
1.1 J.E. Field, Properties of Natural and Synthetic Diamond, Academic Press,
London, 1992.
1.2 G. Davies, Diamond,
Adam Hilger, Bristol, 1984.
1.3 M.N. Yoder in Diamond Films and Coatings, R.F. Davis ed. (Noyes Publications
1993).
1.4 F.P. Bundy, J. Geophys. Res., 85, 6930 (1980).
1.5 J.E. Field, The Properties of Diamond,
Academic, New York, 1979.
1.6 S. Matsumoto, Y. Sato, M. Tsutsumi and N.
Setaka, J. Mater. Sci., 17, 3106
(1982).
1.7 F.G. Celii and J.E. Butler, Ann. Rev. Phys.
Chem., 42, 643 (1991), and
references
therein.
1.8 A.L. Lavoisier, Memoire Acedemie des Science, 564 (1772).
1.9 F.P. Bundy, H.T. Hall, H.M. Strong and R.H.
Wentorf, Nature, 176, 51 (1955).
1.10 W.G. Eversole United States Patent No.
303187 and 303188 (1962).
1.11 R.C. DeVries, Annu. Rev. Mater. Sci., 17, 161 (1987).
1.12 J.C. Angus, H.A. Will and W.S. Stanko, J.
Appl. Phys., 39, 2915 (1968).
1.13 B.V. Spitzyn, L.L. Bouilov and B.V.
Derjaguin, J. Cryst. Growth, 52, 219
(1981).
1.14 S. Matsumoto, Y. Sato, M. Kamo and N.
Setaka, Jpn. J. Appl. Phys., 21,
L183
(1982).
1.15 M.N.R. Ashfold, P.W. May, C.A. Rego, and
N.M. Everitt, Chem. Soc. Rev., 23,
21 (1994).
1.16 J.E. Butler and R.L. Woodin, Phil. Trans.
R. Soc. Lond. A, 342, 209 (1993).
1.17 T.R. Anthony, Vacuum, 41, 1356 (1990).
1.18 D.G. Goodwin, Appl. Phys. Lett., 59, 277 (1991).
1.19 S.J. Harris, J. Appl. Phys., 65, 3044 (1989).
1.20 D.G. Goodwin and G.G. Gavillet, J. Appl.
Phys., 68, 6393 (1990).
1.21 M. Frenklach and H. Wang, Phys. Rev. B, 43, 1520 (1991).
1.22 S.J. Harris, A.M. Weiner and T.A. Perry,
Appl. Phys. Lett., 53, 1605 (1988).
1.23 L.R. Martin and M.W. Hill, Appl. Phys.
Lett., 55, 2248 (1989).
1.24 S.M. Myers and A.F. Zoltowski, Appl. Phys.
Lett., 43, 3425 (1989).
1.25 F.G. Celii and J.E. Butler, J. Appl. Phys.,
71, 2877 (1992).
1.26 F.G. Celii and J.E. Butler, Appl. Phys.
Lett., 54, 1031 (1989).
1.27 M.E. Coltrin, R.J. Kee and J.A. Miller, J.
Electrochem. Soc., 133, 1206 (1986).
1.28 D.N. Belton and S.J. Harris, J. Chem. Phys.,76, 2371 (1992).
1.29 E.H. Wahl, T.G. Owano, C.H. Kruger, P.
Zalicki, Y. Ma and R.N. Zare, Diamond.
Relat. Mater., 5, 373 (1996).
1.30 P. Zalicki, Y. Ma, R.N. Zare E.H. Wahl,
J.R. Dadamio, T.G. Owano and C.H.
Kruger, Chem.
Phys. Lett., 234, 269 (1995).
1.31 S. J. Harris and L.R. Martin, J. Mater.
Res., 5, 2313 (1990).
1.32 S. J. Harris, Appl. Phys. Lett., 56, 2298 (1990).
1.33 M. Frenklach and K.E. Spear, J. Mater.
Res., 3, 133 (1988).
1.34 S.J. Harris and A.M. Weiner, J. Appl.
Phys., 67, 6520 (1990).
1.35 W.L. Hsu and D.M. Tung, Rev. Sci. Instrum.,
63, 4138 (1992).
1.36 S.J. Harris and A.M. Weiner, Thin Solid
Films, 212, 201 (1992).
1.37 W.L. Hsu, Appl. Phys. Lett., 59, 1427 (1991).