The Molecule of the Month --- October, 2004
n-Butane
Recognition of Chemical Bonding Mechanism of Normal Butane Conformers
In Momentum Space
Feng Wang,
FRACI C CHEM
Centre for Molecular Simulation
Faculty of Information & Communication Technology
Swinburne University of Technology
Melbourne, Australia
Introduction
Butane (C4H10) is a colorless,
flammable hydrocarbon that is present natural gas and
can be obtained when petroleum is refined. Butane
is a gaseous alkane. It is extremely stable, has no corrosive
action to metal, slightly soluble in water and readily soluble in alcohol,
ether and chloroform.
Butane is typically used in the manufacture in the following areas
- Aviation fuels and organic chemicals,
- As fuel for cigarette lighters and portable stoves,
- A raw material for synthetic rubber and high octane liquid
fluids,
- Manufacture of ethylene and solvent, propellant in aerosols,
- A calibration gas for temperature and pressure gauges
and as a heating fuel.
- Butane is also added to gasoline in order to increase its
volatility (evaporation rate) in cold climates.
- Recent concerns about the destruction of the ozone layer by
freon gases has led to an increase use of isobutene gas in refrigerating
systems.
Butane exists as two isomers: n-butane is a fully hydrogenated
linear chain of four carbon atoms: CH3CH2CH2CH3
and i-butane, or isobutane, has the formula
CH3CH(CH3)2, and the systematic name
2-methylpropane. Butane is also a protoype in organic
and structural chemistry for structural (constitutional) isomers and conformational
(stereoisomers) isomers. Normal butane is known as n-butane and its chemical
structure looks like
H H H H
| | | |
H - C - C - C - C - H
| | | |
H H H H
Conformational isomers have the same attachment
of atoms but different spatial arrangements caused by rotation about
single bonds [1]. Although conformers are usually difficult
to isolate, in any group of conformers one form is likely to
be more stable than any other, and therefore all molecules of
that compound will spend most of their time in the most stable
conformation. The process of conformational analysis allows one
to predict relative stability of various conformations, and to
gain insight into the behavior of that compound in a chemical
reaction.
Of particular interest and importance are the normal butane conformers
produced by rotation about the central carbon-carbon bond (C(1)-C(2)),
as shown in Figure 1.
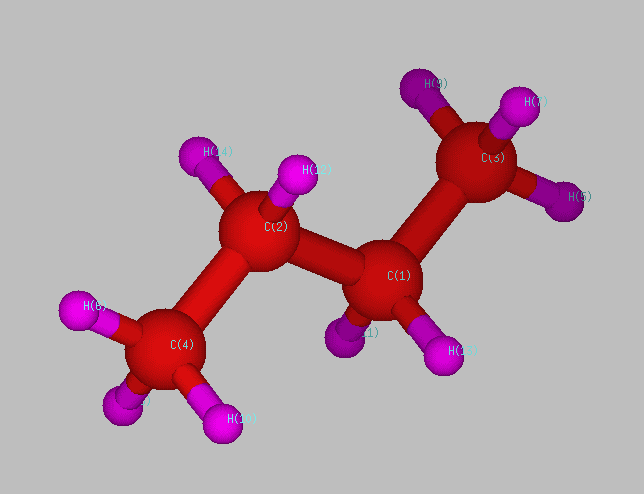
Fig. 1 Normal butane structure
(A).
Conformers of n-Butane
Among these rotationally generated n-butane comformational isomers, we
shall focus on two staggered conformers (A & C) and two eclipsed
conformers (B & D), shown below in Figure 2 as the representable
positions on the rotational potential energy curve [2].
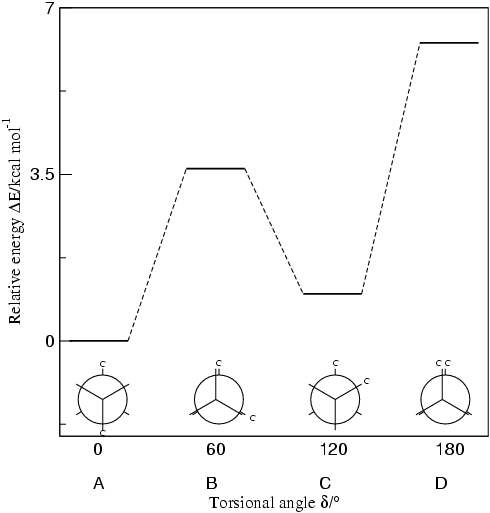
Fig. 2 Rotational energy curve of n-butane calculated by B3LYP/TZVP
model.
The stereo-representation of the butane conformers presented in above
potential energy curve is given below in Fig.3. The staggered conformers
are more stable than the eclipsed conformers by 2.8 to 4.5 kcal/mol. Since
the staggered conformers represent the chief components of a butane sample
they have been given the identifying prefix designations anti
for A and gauche for C [3].
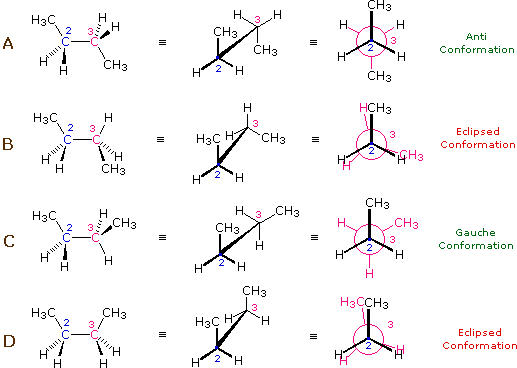
Figure 3 shows the normal butane conformers in several stereo-representations.
As these n-butane conformers exhibit very small energy differences in
their structures, which results in demanding on the experiemental resolution
to observe (when isolation is possible). Theoretical methods such as quantum
mechanical means therefore, plays an important role in the study of conformers
of organic and biological species. In the conventional coordinate space,
however, the energy insensitive properties such as anisotropic (angular
dependent) properties and chemical bonding mechanisms, are challenges to
design new and sensitive experimental means for their observation.
Electron Momentum Spectroscopy and Dual
Space Analysis
Electron momentum spectroscopy (EMS) in conjuction with momentum space
quantum mechanics is able to reveal additional informaton to study orbitals
and chemical bonding mechanisms of molecules [10-11]. Figure 4 demonstrates
how EMS exepriment directly link to binding energy spectrum [12] and individual
orbital momentum distributions (cross sections) [4, 13,14],. And how quantum
mechanics closely works with EMS experiment in both prediction of the
measurable properties and measurement of predictable properties of molecules
and materials.
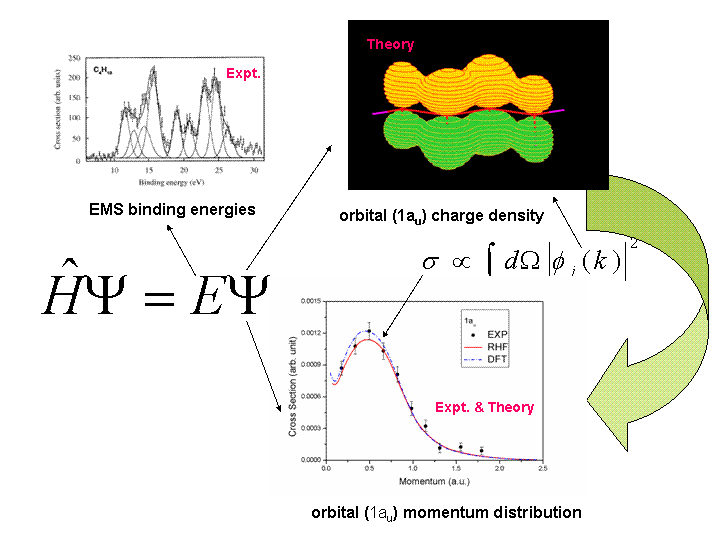
Fig.4 shows how quantum mechnics works with EMS experimental measurements
on n-buatne orbital 1au.
As can be seen from Figure 4 that quantum mechanical studies of molecules
could reach better understanding if electronic information in both coordinate
space and momentum space can be achieved. To this regards, dual space anaylsis
(DSA) [4] was introduced to study bonding mechanism of molecules and to
achieve a more comprehensive understanding of comformers [2,5-6] as well
as tautomers [7] of species without signaficant energy difference. DSA
could also help EMS experiment in distinguishing clustered orbitals in the
outer valence shell due to exeprimental instrumental resolution of the technique.
For example, the three outer most velance orbitals (7ag,
2bg and 6ag) of n-butane cannot be resolved due to
the experimental instrumental resolution
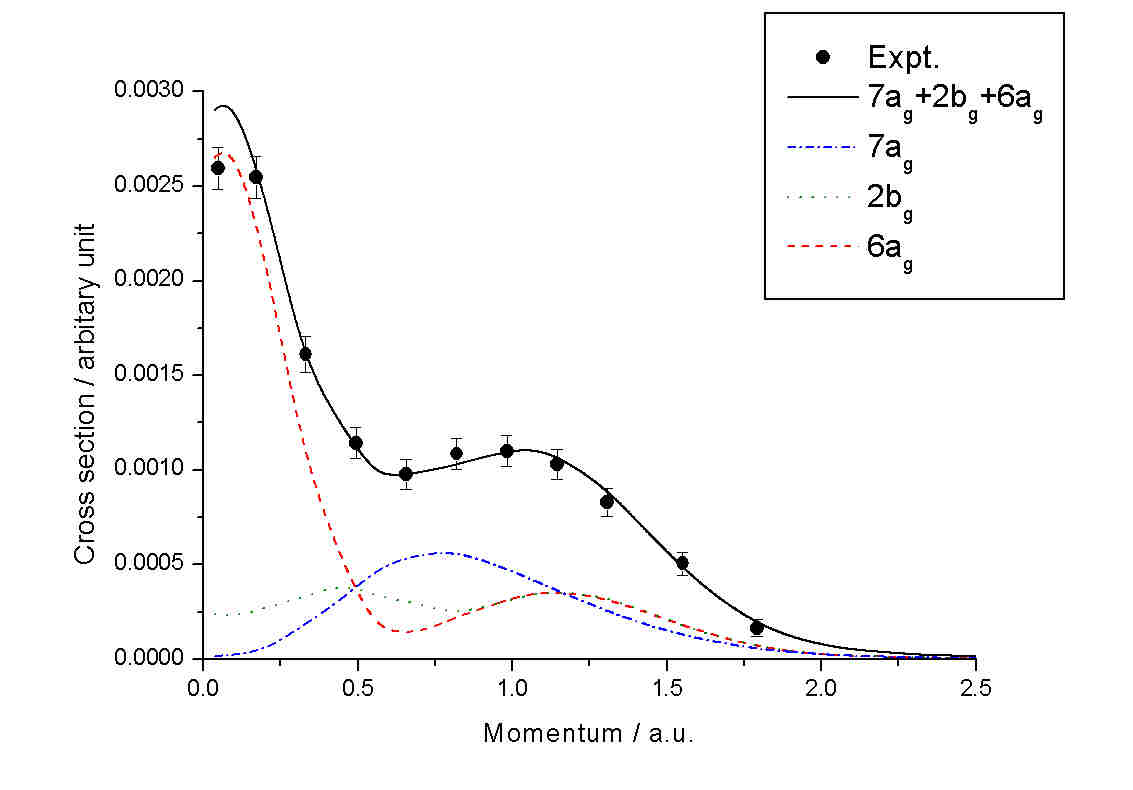
Fig. 5. The outer most cluster of orbitals of n-butane observed
by EMS [11-12, 3] (dots) with theoretically synthesed orbital clusters
and decomposed orbitals.
The highest occupied molecular orbitals (HOMO) of organic molecules
are the target of studies of bonding mechanism and chemical reaction etc.
in organic chemistry since they are very active in chemical processes.
As a result, theoretical DSA method is used to decompose the cluster of
orbitals observed by EMS [4]. The individual orbital momentum distributions
(MDs) of this clustered orbitals of n-butane are shown in the same Figure
with colored curves. It is seen that while exhibit the same point group symmetry,
orbitals 6ag and 7ag have very different bonding characters:
orbital 7ag is strictly p-like but 6ag is formed
by a mixture of s- (smaller momentum region) and p-electrons (larger momentum
region). To arrive a more familiar view of the orbitals, Figure 6 gives
the charge density distributions of the orbitals in coordnate space calculated
using the B3LYP/TZVP model [4].
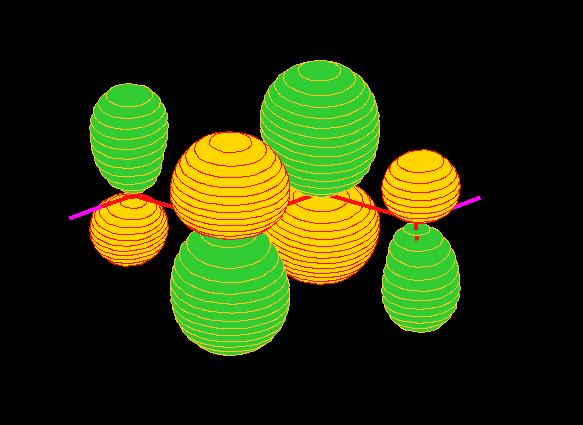

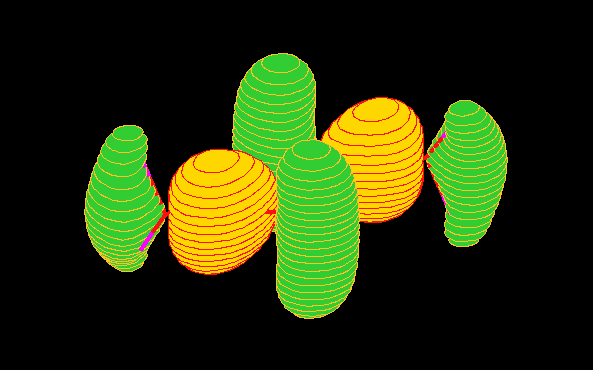
Fig.6 (a) 7ag orbital.
Fig.6 (b) 2bg orbital.
Fig.6 (c) 6ag orbital.
Application to Structure and Bonding Mechanism
Studies
Next question is how could DSA/EMS achieve an insight understanding
of conformers of n-butane? Let's look at the four most significant rotational
conformers of n-butane A (point group symmetry is C2h, ground
electronic state is 1Ag), B(C2, 1A),
C(C2, 1A) and D(C2v, 1A1).
As can be seen from Fig.2, only A and C may be isolated as they represent
the global minimum and local minimum positions on the n-butane rotational
potential energy surface. For example, the outermost orbital, HOMO, of
n-buatne will change when the molecule rotates around its C(1)-C(2)
bond (see Fig. 1). Fig.7 demonstrates such changes in both coordinate space
and momentum space [8],
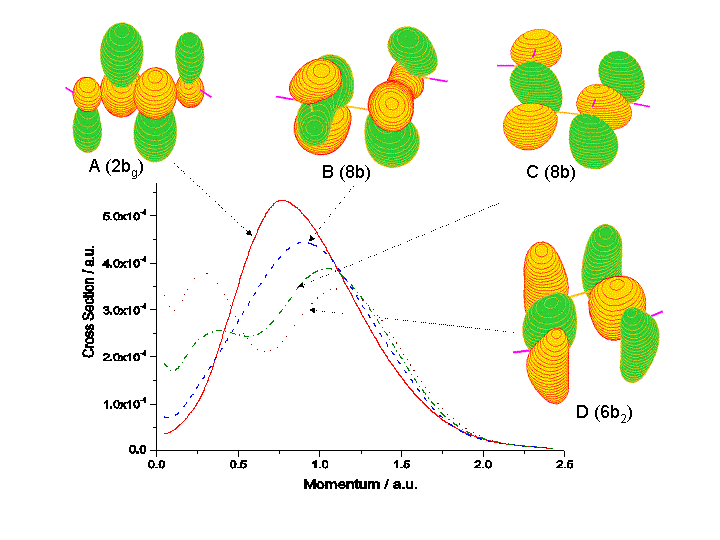
Fig.7 HOMOs of A-B-C-D n-butane in their ground electronic states.
Application to Core Orbitals of n-Butane
Conformers
One of the most significant challenges in computational chemistry
is to extend the prediction power into areas which experiment could not
reach at the moment. To this regard, DSA has been extended into the
core region to study core orbital behaviour of n-butane whent the central C(1)-C(2)
bond is rotating [9]. Because in coordinate space, the information is sensitive
when energy is involved but insensitive when orbitals are in the equivalent
positions in the space, whereas in momentum space, the information such
as equivalent positions in space which cause energy degenerate states and
"invisible" in coordinate space could be significant. For example, the
four core orbitals of n-butane are two-fold degenerate calculated using
RHF/TZVP as shown in Figure 8 [5],
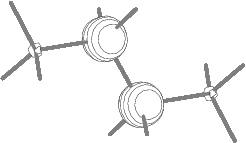
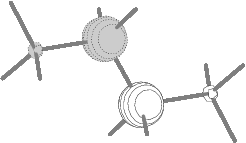
Fig.8(a) Two inner energy
degenerate core orbitals of n-butane
(1ag and 1bu E=305.13 eV).
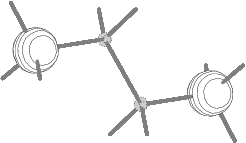
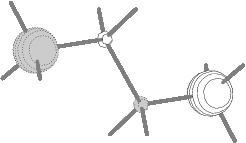
Fig.8(b) Two outer energy
degenerate core orbitals of n-butane
(2ag and 2bu E=304.98 eV).
The orbital momentum distributions of the four core orbitals for the
A, B, C and D conformers are shown in Fig.9 [5],
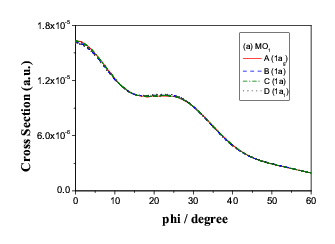
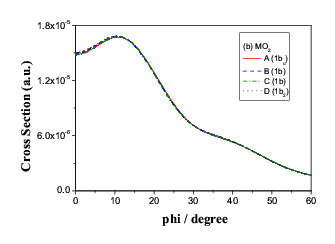
Fig.9(a) Two inner energy degenerate core orbitals of n-butane (1ag
and 1bu)
are also "degenerate" in momentum space.
The inner pair of core orbitals of the four butane conformers does
not change while the molecules rotates around the central bond, C(1)-C(2)
in both coordinate space and momentum space, as the positions of C(1)
and C(2) do not change at all. Only the torsional forces changes
in the rotation. As indicated in Figure 9 (a). However, the C(1)-C(2)
bond rotation causes C(3) and C(4) atoms move to
the equivalent positions in space, as a result, the energy of the orbital
pair exhibits degeneracy but in momentum space, the orbital pair split so
that the conformers can be identified even though the energies are the same.
Figure 9 (b) demonstrate the splitting in momentum space.
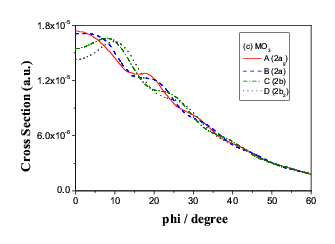
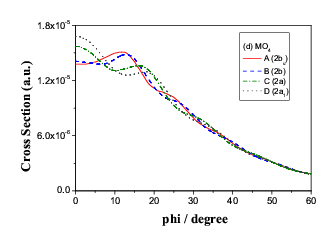
Fig.9(b) Two outer energy degenerate core orbitals of n-butane (2ag
and 2bu)
split in momentum space.
Conclusion
In conclusion, the combined dual space analysis (DSA) and electron
momentum spectroscopy (EMS) could provide an additional dimension (momentum)
of information to assist insight understanding of molecules such as butane,
which has been extensively studied in coordinate space. The information
in momentum space could provide novel information which has been largely
"invisible" in the conventional coordinate space. As conformers and tautomers
largely exist in biological molecules, DSA is expected to play important
role of their structural information anaylsis.
References
- A. Rauk, Orbital interaction Theory of Organic Chemistry, 2nd
ed., Wiley-Interscience, New York, 2001.
- F. Wang and M. Downton,
Inner valence
shell bonding mechanism of n-butane studied using
orbital momentum distributions of its conformational
isomers, Journal of Physics B: Atomic,
Molecular and Optical Physics
37, 557-569(2004).
- See http://www.cem.msu.edu/~reusch/VirtualText/sterisom.htm
- F. Wang, Assessment of quantum mechanical models
based on resolved orbital momentum distributions of
n-butane in the outer valence shell, Journal of Physical
Chemistry A, 107,10199-10207(2003).
- M. Dwonton
and F. Wang, Chemical
bonding mechanisms of n-butane probed by the core molecular
orbitals of conformational isomer in r-space and k-space. Chemical
Physics Letters,
384, 144-149(2004).
- M. Deleuze, W. Pang, A. Salam and R. Shang, J. Am. Chem.
Soc., 123, 4049(2001).
- F. Wang, M. Downton and
N. Kidwani, Adenine tautomer electronic
structural signatures studied using dual space analysis, Journal of Theoretical
and Computational Chemistry, (accepted).
- F. Wang and M. Downton, Orbital topologies and bonding
mechanisms in the outer valence shell of n-butane conformational
isomers (in preperation).
- E. I McCarthy and E. Weigold, Rep. Prog. Phys., 54, 789(1991).
- Weigold and I McCarthy, Electro momentum spectroscopy, Klumer/Plenum,
New York, 1999.
- W. Pang, R. Shang, J. Gao, N. Gao, X. Chen and M. Deleuze,
Chem. Phys. Lett., 296, 605(1998).
- W. Pang, R. Shang, N. Gao, W. Zhang, J. Gao, J. Deng, X. Chen
and Y. Zheng,Phys. Lett. A, 248, 230(1998).
Last updated September 8, 2004

 Back to Molecule of the Month page. [DOI:10.6084/m9.figshare.5437054]
Back to Molecule of the Month page. [DOI:10.6084/m9.figshare.5437054]