Lutein and Zeaxanthin
An Introduction to the Chemistry of Dietary Carotenoids

James D. Johnson
Alumnus, Department of Chemistry
Florida State University, Tallahassee, FL, USA
June 2007
Key: Hyperlink, Pop-up window
;){kind=link}
Introduction:
Carotenoids are fat soluble, colorful pigments serving a variety of roles in cellular biology. Carotenoids are synthesized by plants and microorganisms, primarily photosynthetic. They derive their name from the work of Wachenroder who isolated β-carotene from carrots in 1831 giving the extracted crystals the name 'carotene'. Berzelius (1837) gave the name 'xanthophylls' to more polar pigments having a yellow color (which he extracted from autumn leaves). Richard Willstatter (1907) established the empirical formula of carotenoids (C40) and Tswett (1911), using more advanced techniques in chromatography, separated many pigments which he collectively called 'carotenoids'. Many dedicated scientists in the 20th century, such as Otto Völker of Germany (working with birds), collected and analyzed thousands of pigment samples from hundreds of colorful species, both plant and animal. Today, with the arrival of proteonomics and genomics, the biochemical story of carotenoids is beginning to come into focus, though there is yet much work to be done. Disciplines such as behavioral ecology are poised to take advantage of recent advances in enzymology. The near omnipresence of these pigments throughout the animal and plant kingdoms underlies their importance in a host of biological events. On an evolutionary scale, they are some of the oldest molecules known, originating in the neighborhood of 3 BY ago.
Carotenoids consist of two classes of molecules, q.v., the carotenes, which are strictly hydrocarbons, and xanthophylls, or oxycarotenoids, which contain oxygen. They are predominantly synthesized in nature by photosynthetic organisms and some non-photosynthetic eubacteria. In photosynthesizing species lutein and zeaxanthin, among a few other carotenoids, are integrally associated with the Light Harvesting Chlorophyll Proteins (LHCP) of photosynthetic systems. In plants carotenoids are bound to the trimeric light-harvesting complex II (LHCII, sites L1 and L2). Carotenoids also serve to regulate light harvesting in photosynthetic systems playing a significant role in the organization of thylakoid membranes (allowing LHCP to form trimers, e.g. - thereby increasing the efficiency of light harvesting). Carotenoids are also involved in the distribution of light harvesting complexes between the two photosystems (PSI and PSII). Zeaxanthin, which plays a major role in quenching chlorophyll triplet state energy (if not quenched, can lead to photoinactivation of the photosystems during high light), also quenches singlet oxygen (a highly reactive and toxic form of oxygen). Lutein is essential in the folding of LHCP proteins (bound inside their cores), and, like zeaxanthin, serves as well to quench excess incoming light in the form of chlorophyll triplet state energy. Both lutein and zeaxanthin, along with β-carotene et al., also serve to to transfer incoming light back to the chlorophyll molecules for greater light harvesting efficiency. In this latter capacity carotenoid's serve primarily as light absorbing pigments. Interestingly, the actual transfer of light (referred to as the transfer of an exciton, or 'Energy Excitation Transfer' (EET)), is close or equal to 100% - quite an extraordinary bio-engineering achievement. This makes lutein and zeaxanthin the most abundant carotenoids in nature, and explains why they are found in all vegetative materials, and in most animal species.
;){kind=link}
;){kind=link}
Outside of photosynthesis carotenoids are found in association with biomembranes (generally as an anti-oxidant or membrane stabilizer) or in vesicular domains (storage) such as chromoplasts (flowers, fruits) or oil droplets (avian feathers and eyes).
;){kind=link}
;){kind=link}
Chemically carotenoids consist of isoprenoid building blocks, and are closely related to sterols, ubiquinones, terpenes, Vitamin E (tocopherol), Vitamin K1 (phylloquinone) and a host of other terpenoid compounds (molecules built upon isoprenoid building blocks are collectively referred to as 'terpenoids'). Many of the biosynthetic reactions of carotenoids involve diiron proteins (common in fatty acid desaturases) and cytochrome p450 (common in sterol biosyntheis). Animals may modified dietary carotenoids by further oxidation or cleavage (rarely reduction), but are unable to synthesize carotenoids from basic isoprenoid building blocks.
;){kind=link}
;){kind=link}
;){kind=link}
700+ carotenoids have been identified to date. On a species by species basis only a few carotenoids are generally acquired and/or synthesized (generally < 10). Many colorful songbirds, e.g., endogenously modify incoming dietary carotenoids, such as lutein and zeaxanthin, into six colorful red and yellow pigments (they can synthesize additional red pigments using β-carotene and β-cryptoxanthin, also considered dietary carotenoids). Many species have only a few specialized carotenoids, such as generally found in bacteria. Lutein and Zeaxanthin are considered dietary carotenoids because of their occurence in vegetative material and their requirement in animal physiology.
In higher plants zeaxanthin is a precursor and regulator in the synthesis of abscisic acid (ABA), a plant growth hormone synthesized in response to water stress. In animals, the oxidative cleavage of dietary β-carotene, a-carotene, and/or β-cryptoxanthin, a xanthophyll, to yield Vitamin A. In the last 20 years the importance of carotenoids in the diet of humans has been linked to the prevention of age-related macular degeneration (ARMD) and age-related cataract formation (macula of the eye contains lutein and zeaxanthin). Carotenoids have also been implicated in anti-cancer activity, increased immunocompetence and general anti-oxidant protection of metabolic activity (take home message, eat plenty of fresh green vegetables!).
;){kind=link}
;){kind=link}
General Properties of Carotenoids
Carotenoid behavior is derived substantially from their highly delocalized polyene backbone. This system is further exploited by two cyclohexene end rings. Depending on the degree of substitution, these rings can strongly influence the overall properties of the carotenoid. Lutein, which is a soft pastoral yellow, can be ketonized (C-H ![]() C=O) at the fourth ring carbon resulting in an extended (longer) conjugation of double bonds with the cartotenoid polyene backbone. This causes the spectrum of lutein to shift to the red (bathocrhomic shift). Oxidation of the end rings also influences their ability to act as anti-oxidants, in many cases increasing their capacity to quench both chlorophyll and radical oxygen species. Chemical protection is also afforded through oxidation of the end rings thereby preventing radical attack on unconjugated ring double bonds or hydroxyl groups. Radical oxidation often leads to destruction of the carotenoid, commonly referred to as 'sacrificial radical scavenging' (more on this below).
C=O) at the fourth ring carbon resulting in an extended (longer) conjugation of double bonds with the cartotenoid polyene backbone. This causes the spectrum of lutein to shift to the red (bathocrhomic shift). Oxidation of the end rings also influences their ability to act as anti-oxidants, in many cases increasing their capacity to quench both chlorophyll and radical oxygen species. Chemical protection is also afforded through oxidation of the end rings thereby preventing radical attack on unconjugated ring double bonds or hydroxyl groups. Radical oxidation often leads to destruction of the carotenoid, commonly referred to as 'sacrificial radical scavenging' (more on this below).
When carotenoids are synthesized for the purpose of absorbing visible light they are said to behave as chromophores. The red of a rose pedal, or the color of fruit, or of the beautiful displays in songbirds are good examples. The use of carotenoids for colorful diplays in animals is the result of further oxidation of dietary carotenoids (lutein and zeaxanthin) typically invoked as a mechanism to enhance dichromatic behavior between the sexes. Chromophoric carotenoids are also found in oil droplets within the retina of birds and reptiles, serving the purpose of cut-off filters comparable to those found in cameras. The result is an enhanced sharpness of percieved colors (birds are tetrachromatic). [Note: While birds sport tetrachromatic color vision, the loss of high resolution color vision in mammals can be attributed to birds and their relatives. It was the dinosaurs that drove mammals into a nocturnal underground habitat, with the subsequent evolutionary loss of high resolution daytime color vision].
Carotenoids also have the ability to 'vibrate away' their triplet state energy as heat into the surroundings. This enables carotenoids to absorb ('quench') the energy of excited molecules such as singlet state oxygen and triplet state chlorophyll, thereby preventing the destruction of protein, DNA and the photosynthetic apparatus. In photosynthetic tissue, carotenoids play a vital role in the organization of each photosystem, regulating incoming and outgoing light intensity. Carotenoids are very efficient at absorbing and transmitting exciton energy (EET as defined above) when and where needed. Carotenoids can re-emit an exciton to a neighboring chlorophyll molecule (thereby broadening the photosystem's absorption spectrum or 'antenna size'), or can harmlessly release the energy packet as heat through vibration (see below). Finally, a carotenoid may serve as a sacrificial molecule in which reactive radicals chemically attack it. Through its anti-oxidant behavior carotenoids protect against the peroxidation of phospholipids (Lipid peroxidation), a chain reaction, as well as proteins and DNA. Like sterols carotenoids may also play a role in controlling membrane fluidity.
;){kind=link}
The Primary role of Carotenoids in Photosynthetic membranes
β-carotene can be found in close association with PSII, located adjacent to the reaction center (see image below, left). β-carotene plays a dual role at the reaction center, providing incoming photons to the reaction center (singlet to singlet transfer) or quenching excess light in the form of chlorophyll triplet state energy transfer (subsequently releasing it as heat). β-carotene is the only carotenoid bound to the core reaction center complex of photosystem II. Lutein, the most abundant carotenoid in nature, is bound to two sites (L1 and L2) inside the core of PSII's LHC II chlorophyll-protein complex. Lutein is the most abundant carotenoid in thylakoids, and plays an important role in quenching chlorophyll triplet state and singlet state oxygen during times of maximum daylight when the reaction center is saturated. In the latter 'high stress' state carotenoids are said to be part of the NPQ (non-photochemical quenching) pool of anti-oxidants (feedback de-excitation). In addition lutein is required for proper LHC II folding and organization. Zeaxanthin [MDL] is associated with photo-regulation of the photosynthetic process in close association with the LHCP. Zeaxanthin also plays a secondary role in preventing lipid peroxidation througout the thylakoid membranes.
;){kind=link}
;){kind=link}
;){kind=link}
Zeaxanthin is considered a regulator of light harvesting, and plays a fundamental role in the Xanthophyll Cycle of PSII. Zeaxanthin carries out three functions during high light conditions, q.v., (1) protection against photo-oxidation due to radical oxygen attack (physical quenching of oxygen singlet energy), (2) absorption of chlorophyll triplet energy (which, if not captured, leads to singlet oxygen formation through a process in which ground state triplet oxygen absorbs chlorophyll triplet energy becoming a dangerously reactive oxygen singlet species). Captured singlet state energy of oxygen by the carotenoid is subsequently lost as vibrational heat into the surrounding medium (due to the high level of conjugation of carotenoids molecule it is able to vibrate and release heat in this fashion), and (3) to absorb incoming photons and to transfer these photons to neighboring chlorophyll molecules thereby increasing the overall absorption spectrum of the photosytem in which it is participating (carotenoids account for ~20-30% of all light harvested).
;){kind=link}
Below are two images showing the presence of carotenoids at the very heart of photosynthesis. β-carotene can be seen close to the reaction center of Photosystem II (left), and rhodopin glucoside (an acyclic carotenoid with a sugar attached), associated with the Light Harvesting Chlorophyll Protein 2 (LH II) of Rhodopseudomonas acidophila, a photosynthetic purple bacteria (right). The blue line on the oxidizing side of PSII (left) depicts the path of electrons from water, located at the site of the manganese cluster (the Oxygen Evolving Complex or OEC), to quinone, which captures the electrons for processing in the electron transport chain.
;){kind=link}
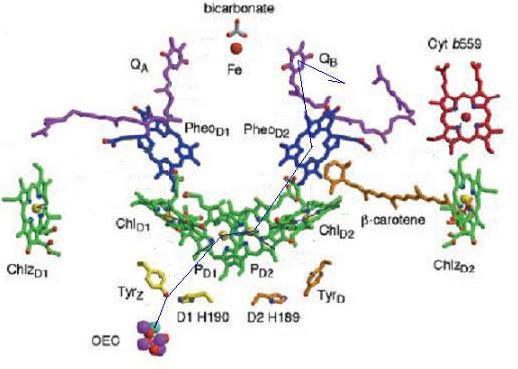 |
Carotenoids may also serve as conductors of electrons. Carotenoids possess a chain of alternating double and single C-C bonds suggesting that they can act as ‘‘molecular wires ’’. In isolated photosystem II preparations carotenoids appear to have the ability to conduct electrons from cytochrome b559 to the reaction center P680 chlorophylls. The exact nature of the process has not been determined in vivo but there is little doubt that a molecule of β-carotene is able to conduct electrons over a distance of ~30 angstroms.
A few Notes on the chemical structure of Carotenoids
Carotenoids may vary in chain length, but the majority contain forty carbon atoms (C40). Carotenoids are highly conjugated polyene chromophores (9-13 double bonds, the most common appearing as all-trans), and it is this conjugation which gives carotenoids much of their functionality. The large number of isomers possible with carotenoids, considering cis/trans configurations in conjunction with chiral centers, can lead to tens of thousands of potential combinations. The requirement for planarity (conjugated system) substantially restricts this isomeric potential. Although there are a few stable cis molecules, their formation is generally prohibited by steric hindrance of methly groups during rotation. Several enzymes specifically bind cis-carotenoids (such as 9-cis-epoxycarotenoid dioxygenase) but in general cis-carotenoids are uncommon in biological systems.
;){kind=link}
Biological requirements, such as membrane positioning, also plays a role in restricting isomers, especially cis/trans. Also restricting the potential number of isomers is the pervasive keto-enol tautomerization that is found in the modified ring systems of carotenoids. In an example given below, a single carotenoid end-product, using keto-enol tautomerization, is shown to have six potential isomers. Steric hindrance will further reduce these isomers to ones thermodynamically and sterically favored. As is true in many areas of carotenoid chemistry, the most stable isomer is generally the one in which the resulting keto group is in resonance with the polyene chain (thereby restricting the appearance of alternative tautomers - in at least one bird species a non-resonance tautomer was found at 6% levels in feathers perhaps favored by the keratin matrix in which it was despoited). When carotenoids are cleaved, or fragmented, they are referred to as 'apocarotenoids'. Apocarotenoid formation may be non-specific (lipoxygenase, photo-oxidation, radical oxidation) or may be the result of specific enzymatic reaction such as occurs in the dioxygenase family (examples of cleavage products).
;){kind=link}
Carotenoids are numbered according to the following scheme:
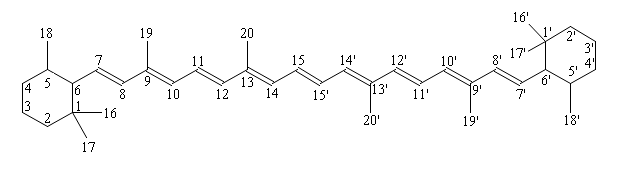
β-carotene
Carotenoid: IUPAC Definition
Carotenoids are a class of hydrocarbons (carotenes) and their oxygenated derivatives (xanthophylls) consisting of eight isoprenoid units joined in such a manner that the arrangement of isoprenoid units is reversed at the center of the molecule so that the two central methyl groups are in a 1,6-positional relationship and the remaining nonterminal methyl groups are in a 1,5-positional relationship. All carotenoids may be formally derived from the acyclic precursors, having a long central chain of conjugated double bonds [enzymatically produced by] (i) hydrogenation, (ii) dehydrogenation, (iii) cyclization, or (iv) oxidation. or any combination of these processes" (IUPAC Carotenoid Nomenclature Rules).
A ball and stick model of lutein (an 'oxycarotenoid' or 'xanthophyll') is shown below.
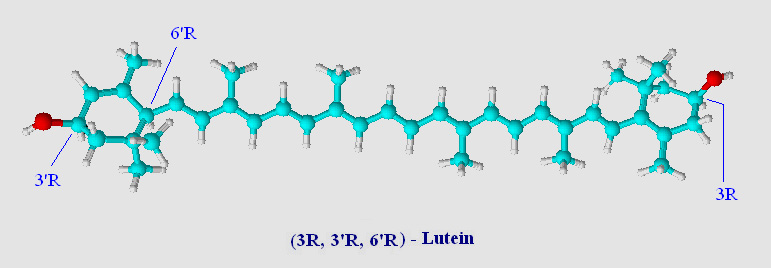
Note that the two rings have been hydroxylated at the 3 and 3' positions. Chemists call the two rings of lutein beta (β, double bond between C5=C6) and epsilon (ε, double bond between C4=C5). The location of a single double bond is the only difference between lutein and zeaxanthin (visual comparison). Lutein has one β-ring and one ε-ring, zeaxanthin has two β-rings. Chemically lutein and zeaxanthin are isomers, although not stereoisomers, and occur naturally as all-trans (all-E - note in carotenoids trans is synomous with E unless there are oxygens substitutions on the carotenoid chain) geometric isomers (see Virtual Organic Chem I and II for a brief overview on organic isomers including R,S and E,Z terminology).
;){kind=link}
Lutein has three chiral-carbons which are enantiomeric (optically active). These centers are shown in the above image located at C3, C3' and C6' and are designated as S (sinister as compared to R, rectus) (see Cahn-Ingold-Prelog priority rules). Normally this would be a blessing to biochemists, giving stereochemical handles on a carotenoid prior to assessing catalytic activity. Stereochemical centers can lead to important information on the stereospecificity of catalytic events. With carotenoids however the number of stereoisomers is a mixed blessing. Oxidized carotenoid rings show a high degree of tautomerization (a form of keto-enol isomerization), and a substrate or product can often times be represented by several tautomeric isomers. In many cases tautomeric isomers of carotenoids have low energy barriers separating them from R and S configurations. As a result, often times these stereocenters are lost in the shuffle so far as an initial reference point may be defined. Fortunately, the high degree of conjugation in carotenoids tends to favor those tautomers which are in resonance with the backbone of the carotenoid. Several sterol enzymes have been found to assist in keto-enol tautomerization (enzyme assisted isomerisation) favoring one isomer over another.
In keto-enol or tautomeric isomerisation the keto form is generally the most stable form thermodynamically. To better appreciate the high degree of tautomer isomerisation in carotenoids take the time to work out the following exercise.
Exercise: Search for Tautomers
- Keto-enol tuatomerization: The Rules
- Sketch this structure on paper. This 'half carotenoid' represents, e.g., an enzyme product.
- Find (1) the most stable tautomer (and expected product, represented by R and S forms - Hint: most stable is in resonance with the backbone of the molecule) as well as the remaining four (4) tautomeric isomers that exist for this product (all tautomers are inside the ring).
- [Solution]
;){kind=link}
;){kind=link}
Many times a compound is overlooked, or an isomer identified as unique, when tautomers (keto-enol isomerisation) are involved. Recognizing an allylic ketone or an alcohol attached to a desaturated carbon should invoke a thorough search for the various isomers, making sure to keep track of any chiral centers and their behavior. Generally the lowest energy tautomer, a ketone, is spontaneously generated since it is favored thermodynamically. In carotenoids we find that the most stable tautomers are those in which the ketone is ultimately in resonance with the existing conjugated system. In one case of carotenoid tautomer search undertaken by this author it was found that a double bond was relocated from one side of the ring to the other, involving at least a half a dozen intermediates along the way.
Generally carotenoids are found in all-trans conformation due to the requirement that planarity is required for maximum pi-electron delocalisation (the carotenoid is also at its lowest energy). With a slight 'S' angularity across the backbone and minor rotation at the ring interface, both to relieve steric strain, the majority of carotenoids achieve a highly functional delocalized system. When cis isomers occur they are generally found at positions 9, 13 and 15. It is as these positions that CH3 (methyl) + H (hydrogen) steric hindrance is minimal (review the numbering image above). When looking at the ring-chain junction, 6(S)-trans isomer is preferred over 6(S)-cis isomer although a 40 degree angle from planarity is necessary to offset steric hindrance between the C5 methyl and C8 hydrogen (see cis-trans comparisons).
;){kind=link}
A few Notes on Carotenoid Chemistry
A carotenoid's polyene chain is susceptible to radical attack primarily because the delocalisation of pi-electrons makes this region of the molecule electron rich. Electron density maps have shown that the highest density occurrs near the end of the chain (positions C7 through C9) or on the end rings if oxygenated (electron withdrawing groups are present). For example, chain cleavage reactions (oxidations producing apocarotenoids, e.g.) tend to favor the C7-C8 position of the carotenoid, the position of the final double bond on the backbone chain (biologically cleavage reactions are typically carried out by diooxygenases, such as the one that produces retinal from β-carotene). The highly delocalized electrons of carotenoids make them quite suitable for stabilizing reactive intermediates such as carbocations or radicals. The energy required to extract a hydrogen atom (homolytic cleavage) is substantially lowered if the proton is allylic e.g., a proton extraction on C3 of a β-ring is lowered by ~ 15 kcal/mol. I was unable to find the energy required to extract the C4 hydrogen from a β-ring in full conjugation wtih the carotenoid backbone but would be expected to be significantly less then the previous example. The ease of homolytically extracting the C4 hydrogen of a β-ring makes it an ideal location for radical attack (the resulting radical is significantly stabilized thereby making the extraction an easy target). In many cases oxygen singlet state species prefer to attack this position because of its long lifetime and preference for low energy interactions.
;){kind=link}
The image below has been included to show the various relationships between a carotenoid and its immediate environment (using zeaxanthin and its relationship with PSII's LHCP). Note: The chemistry of carotenoids and radicals is somewhat complex and has been shown to be highly dependent on the environment in which the carotenoid is found. The image below depicts some of the events which occur within the thylakoids of photosynthetic membranes. Anti-oxidant behavior can be demonstrated, e.g., by measuring the spin state signature of radical species. For a reveiw of carotenoid radcial chemistry see the reference section below.
;){kind=link}
Consider first the left side of the image below, labeled 'anti-oxidant'. Carotenoids serve as anti-oxidants in one of two ways. They can physically "quench" singlet oxygen energy (thereby de-fusing the highly reactive species back to its triplet ground state) or scavenge free radicals by chemically reacting with them forming a new carotenoid radical (this pathway is sacrificial since the carotenoid is generally cleaved or made inoperable in the reaction). Chlorophyll is shown absorbing incoming light creating a chlorophyll singlet excited state. From this state chlorophyll may loose a small amount of energy converting it to its lower energy triplet state. If the triplet state is left unchecked it is l;ikely to create singlet oxygen from triplet ground state oxygen. Generally in this situation chlorophyll's triplet state energy is transferred through intersystem crossing to a nearby carotenoid, which is then left in its excited triplet state. Since the carotenoid's triplet state energy is low, it has only one path to release the energy, q.v., as vibrational heat into the medium. The left side of the image below depicts the two pathways for chlorophyll triplet energy transfer. The carotenoid can absorb this energy from either triplet chlorophyll or singlet oxygen. If a carotenoid is not nearby to accept oxygen's excess energy (or is not properly oriented to do so) then it will oxidatively attack nearby lipids, proteins or even nucleic acids (mRNA, tRNA and/or DNA).
Note in the section labeled 'Increase photosystem (PS) cross section' zeaxanthin absorbs a photon of visible light (designated by the carotenoid's transfer from its ground state (So) to its second singlet state (S2)), and transfers this energy to a nearby chlorophyll thereby increasing the overall photo-cross section of the photosystem (broadens the spectrum of the PSII unit thereby enhacing overall antennae light harvesting efficiency). In the image above showing β-carotene close to the reaction center of PSII, a singlet energy transfer of a photon occurs between the carotenoid and Chlorophyll a, or, alternatively, chlorophyll triplet energy is sent to the carotenoid. In at least one case the exchange of triplet energy between carotenoid and a nearby chlorophyll appears to be mediated by oxygen. In the area marked 'sacrificial radical scavenging' at the bottom of the image carotenoids are shown to be consumed by direct chemical attack of a radical species if intersystem transfer of excited energy to the carotenoid is unsuccessful (e.g., the carotenoid is not properly oriented, usually this is accomplished via protein binding). Note that a line drawn from chlorophyll triplet excited state to its ground singlet state represents chlorophyll fluorescence. In many cases of radical carotenoid chemistry electron transfer takes place between carotenoids and other reactant molecules (radical cation/anion chemistry).
;){kind=link}
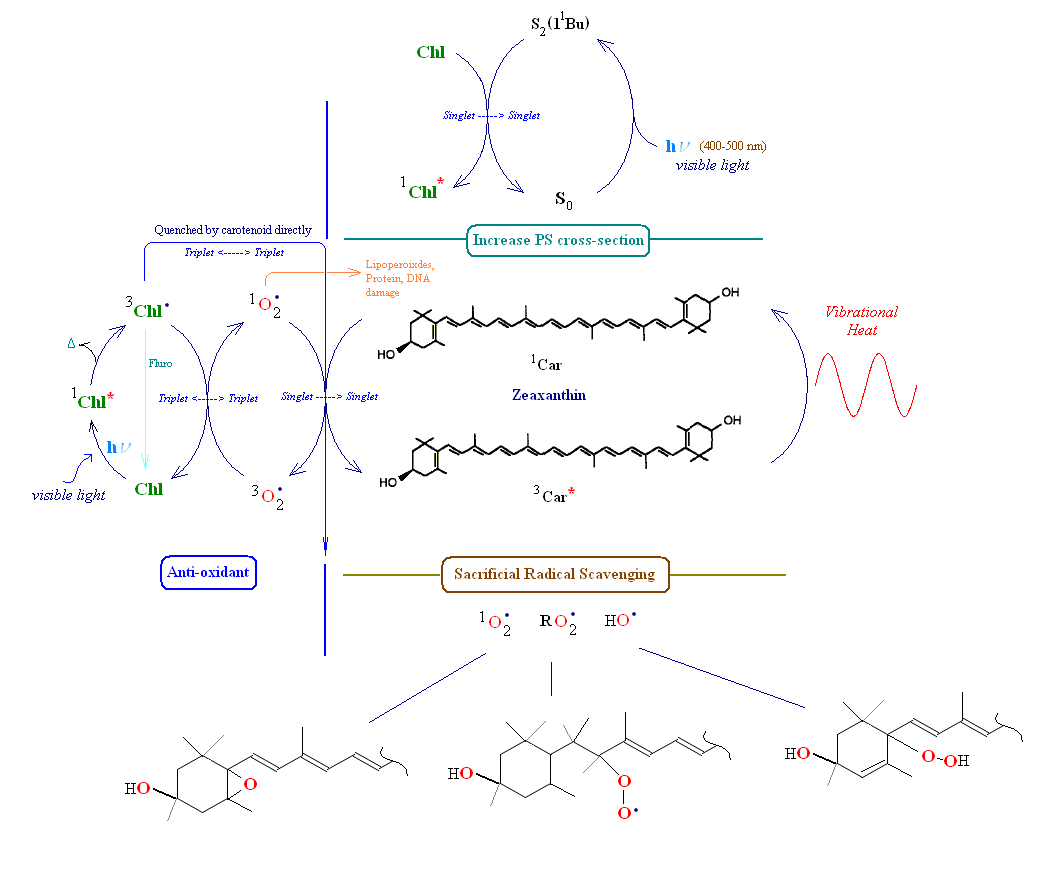
Highly delocalized electrons in carotenoids are primarily responsible for their rapid oxidative degradation following exposure to even minor levels of oxygen (e.g., after purification). In cellular compartments and membranes carotenoids are protected against oxidation through association with proteins, fats and other nearby molecules. Even with inherent biological protection carotenoids are susceptible to radical attack (especially by superoixdes and peroxides). In many oxidation reactions the carotenoid is cleaved into smaller fragments (creating apocarotenals and apocarotenones). In others epoxides and peroxide radicals are formed. When a carotenoid confronts singlet oxygen and intersystem transfer of oxygen's excess energy to the carotenoid is unsuccessful, singlet oxygen may directly attack the carotenoid (shown above as 'sacrificial savenging'). This latter reaction changes the carotenoid from an anti-oxidant to a pro-oxidant. Sacrificial scavenging may also lead to a break up in a local lipid peroxide chain reaction. If the carotenoid is cleaved in the oxidation process, the resulting apocarotenoids are quickly further broken down into soluble metabolites.
One more example is shown below in order that the reader better understand the balance between a carotenoid behaving as an anti-oxidant by quenching singlet oxygen, or result in sacrificial scavenging. Notice that those rings (e.g., β-carotene) which have exposed C4 hydrogens, are most likely to become oxidized by radical species themselves (lower energy to extract since resulting radical will be in resonance). Therefore, those species shown below that are least susceptible to radical attack are those that protect the low energy hydrogens at the C4 position (always start with the dimethyl group as C1, then counter-clockwise to C6). The last carotenoid shown below, with a hydroxyl at the C2 position (rare), is the strongest anti-oxidant quencher. The presence of a 4-keto-3-hydroxy group on the end rings (astaxanthin) greatly enhances the ability for intersystem transfer (hence increases the rate at which a carotenoid can react with singlet oxygen). Interestingly, and perhaps related to polar-polar interaction with the end groups, a rare C2 hydroxyl group has been found to impart unusually high intersystem transfer capabilities to the carotenoid. The quantum behavior of carotenoids as a function of ring substitution is currently an active area of research in anti-oxidant biochemistry.
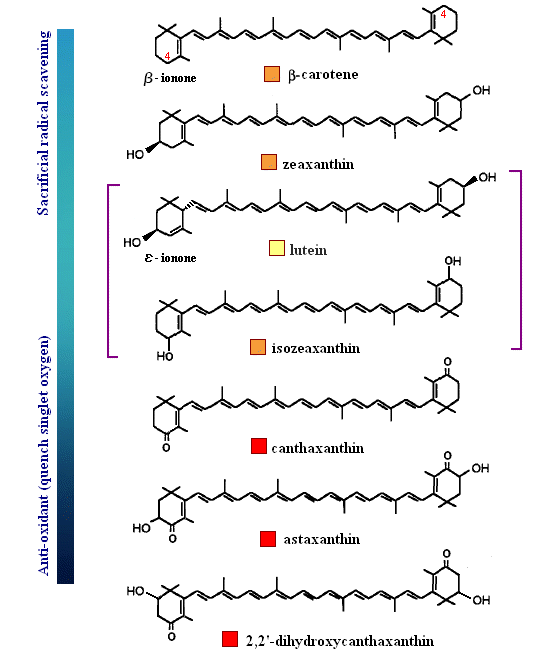
Notes on the Biosynthesis of Carotenoids
The relationship between the biosynthesis of carotenoids and other terpenoid biomolecules is shown below.
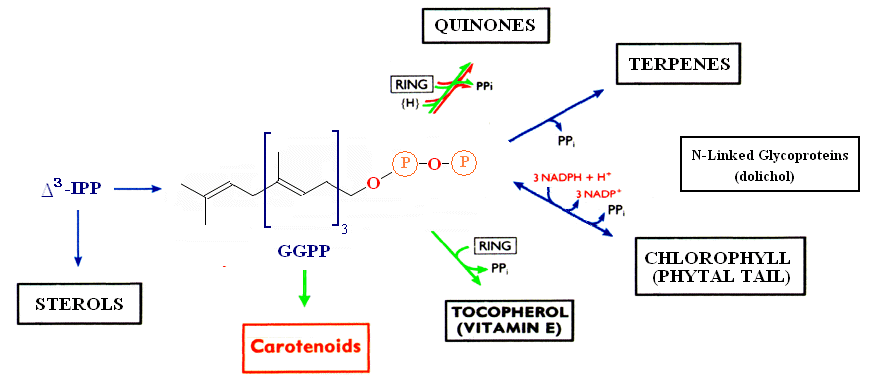
The metabolite common to all classes of molecules within the terpenoid pathway is Isopentenyldiphosphate. IPP serves as the precursor for several terpenoid molecules, including the phytal chain of chlorophylls, provitamin A and Vitamin E (α-tocopherol), quinones, terpenes, gibberrellins and accessory photosynthetic pigments. For an excellent overall review of Carotenoid biosynthesis see MetaCyc's metabolic pathways. The principle pathways leading to the most common carotenoids are shown here (note that β-carotene also serves as a precursor to Vitamin A).
;){kind=link}
;){kind=link}
;){kind=link}
;){kind=link}
;){kind=link}
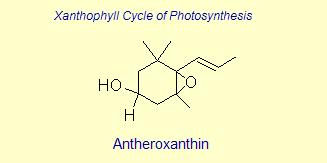
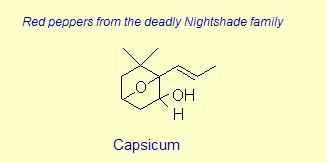
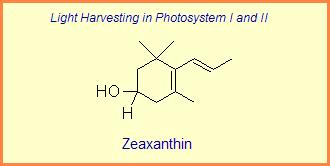
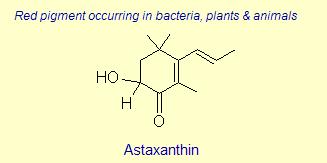
Diversity of End Rings in Naturally occuring Carotenoids
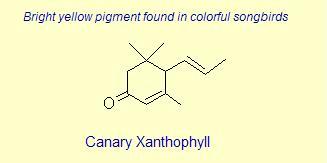
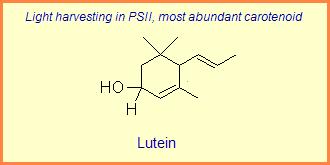
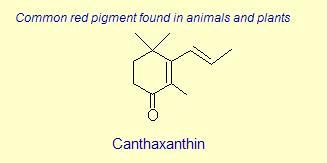
The variation of themes found in carotenoids across the biosphere is quite remarkable. The primary dietary carotenoids, which includ lutein, zeaxanthin, β-carotene and β-cryptoxanthin are abundant in most plant tissue and serve primary roles in photosynthesis, hence their omnipresence throughout the plant kingdom. More specialized carotenoids, synthesized by both plant and animals, is primarily the result of biosynthetic modification of the end rings of dietary carotenoids. These modifications tend to convert the pastoral yellows and light orange colors of dietary carotenoids to red and yellow pigments (as a result most of the yellows, oranges and reds in nature are carotenoids). It would require another web page to even begin to describe the diversity that does occur, so a few selected end rings are presented below in order for the reader to better appreciate some of the chemical architecture that occurs in carotenoids. Serveral of the general references listed below have summary lists of carotenoids if one is interested in seeing a database of carotenoid types.
 |
 |
 |
 |
 |
 |
 |
 |
 |
Carotenoid Enzymes
Dietary carotenoids such as Lutein and Zeaxanthin are subject to several modifying enzymes that typically further oxidize the carotenoid. In animals dietary carotenoids such as β-carotene serve as precursors to retinal, which is bound to rhodopsin for vision. Many species create colorful pigments used as indicators of good health and immunocompetence during mate selection (these tend to be red and yellow pigments, or combinations of the two). Further oxidation also increases the rate of singlet oxygen quenching, and so the production, e.g., of astaxanthin may be advantageous to the organism for protection against lipid peroxidation or other biochemical oxidative damage. Furthermore enzymes replace biological active carotenoids (or obtain new ones if dietary) if they are sacrificed, e.g., through radical scavenging. Carotenoids are subject to a host of modifying reactions, including desaturation, oxygenation, hydrogenation, isomerization, cyclization and oxido-reduction. A few of the most common carotenoid enzyme types found in plants and animals are discussed briefly below.

Non-heme diiron.The diiron family consists of soluble and membrane bound desaturases/hydroxylases. All non-heme diiron enzymes are capable of carrying out a desaturase and/or hydroxylase reaction. Members of this family of proteins include the oxygen carrier Hemerythrin (oxygen carrier in marine invertebrates), methane monooxygenase (converts methane to methanol in methanogenic organisms), ribonucleotide reductase and purple phosphatase. Membrane bound diirons typically work in conjunction Cytochrome b5 and Cytochrome b5 oxido-reductase (fatty acid desaturases). Evolutionarily these enzyme closely associated with both fatty acid metabolism and sterol biosynthesis. There is evidence that diiron enzymes were critical as newly released oxygen began to enter the biosphere at the time of the rise of the Cyanobactera and the oxygen evolving complex of PSII. Diiron enzymes have diverged to many varities (with little homology between them) but have retained a stictly conserved 3 Tier Histidince motif, necessary for liganding to the two catalytic iron centers (H-H-HXXXH-HXXHH, where H is Histidine). The enzymes are quite versatile, and may be biochemically modified to favor desaturation over hydroxyation with a common substrate. Small changes in enzyme behavior in vivo have been shown to be sensitive to several highly conserved, but not obligatory, amino acids that appear to be similar to the Structural Recognition Sites (SRS) on P450s, that is, they are mild adjusters of 'docking conformity'. Membrane physiology may also play a role in the behavior of these enzymes, catalytic activity may be favored when the bilipid membrane is in 'top' condition (requisiste phospholipids). Diirons, like P450s, use hydrogen abstraction (radical homolysis) followed by either hydroxylation, aka 'oxygen rebound' (monooxygeanse) or a second hydrogen extraction (desaturase). Diiron enzymes require associated oxido-reductase systems such as NADPH or NADH in order to supply electrons to activate incoming oxygen. In many cases, associated oxido-reductase systems cooperatively enhance enzyme catalytic rates. The oxido-reductase will depend on whether the diiron is soluble or membrane bound. NADH and oxygen are consumed in each cycle of the enzyme.
;){kind=link}
In the above image of the soluble diiron (δ-9 Desaturase) the two catalytic iron atoms can be seen centered within a 4-alpha helical trans-membrane bundle. A view from the top of this enzyme is seen in the middle image. The image on the right depicts the substrate binding environment. Not much is known about the catalytic site for integral membrane bound diirons, they are not soluble and so are not easily isolated. Many mutational studies have been done to date on membrane bound diirons revealing their regio- and stereochemical selectivity.
 Class II Cytochrome p450. It is a remarkable result that both diiron non heme enzymes and Cyctochrome p450 enzymes share many similar catalytic features. Both use the oxygen-rebound monooxygenase and desaturase reactions. Both require oxygen to activate the iron and both are associated with electron dontaing reductases (involving NADPH and FAD - cf p450 mechanism). Both are found catalyzing carotenoids in various species. As a general rule of thumb p450 prefers a frontal approach to its substrate oxygenating C3 on carotenoids rather then C4 (at least one C4 oxygenase has been found). Sterols use p450 for most of their oxygenase reactions with a few exceptions (e.g., a diiron is used for the conversion of cholesterol to 25-hydroxy-cholesterol). Class II Cytochrome p450s are found within the endoplasmic reticulum. p450 is anchored into the cytoplasmic side of the bilayer with a 20-30 amino acid anchor (N terminal). Diirons and p450s are collectively referred to by biochemists as 'cellular blow-torches' because of their ability to homolytically cleave C-H bond and convert them to ketones (C=O). In order for a carotenoid to enter the active site of p450 it must be partitioned completely out of the membrane and up into the binding site of the p450 (see example here - look closely for the carotenoid, colored grey. The carotenoid is shown nearly completely removed from the bilayer with the non-reacting ring interacting with the phospholipids at the surface - this behavior is also seen in Cholesterol oxidase, a rare oxido-reductase enzyme carrying out both an oxidation and isomerization reaction. In the image of the p450 complex (left) a carotenoid is shown docked at the catatlyic site. Several critical catalytic site amino acids (both polar and hydrophobic, designated as "SRS" or substrate recognition sites) interact closely with the docked carotenoid. The C4 carbon of the carotenoid is approximately 4 Angstroms above the activated iron-oxygen heme.
Class II Cytochrome p450. It is a remarkable result that both diiron non heme enzymes and Cyctochrome p450 enzymes share many similar catalytic features. Both use the oxygen-rebound monooxygenase and desaturase reactions. Both require oxygen to activate the iron and both are associated with electron dontaing reductases (involving NADPH and FAD - cf p450 mechanism). Both are found catalyzing carotenoids in various species. As a general rule of thumb p450 prefers a frontal approach to its substrate oxygenating C3 on carotenoids rather then C4 (at least one C4 oxygenase has been found). Sterols use p450 for most of their oxygenase reactions with a few exceptions (e.g., a diiron is used for the conversion of cholesterol to 25-hydroxy-cholesterol). Class II Cytochrome p450s are found within the endoplasmic reticulum. p450 is anchored into the cytoplasmic side of the bilayer with a 20-30 amino acid anchor (N terminal). Diirons and p450s are collectively referred to by biochemists as 'cellular blow-torches' because of their ability to homolytically cleave C-H bond and convert them to ketones (C=O). In order for a carotenoid to enter the active site of p450 it must be partitioned completely out of the membrane and up into the binding site of the p450 (see example here - look closely for the carotenoid, colored grey. The carotenoid is shown nearly completely removed from the bilayer with the non-reacting ring interacting with the phospholipids at the surface - this behavior is also seen in Cholesterol oxidase, a rare oxido-reductase enzyme carrying out both an oxidation and isomerization reaction. In the image of the p450 complex (left) a carotenoid is shown docked at the catatlyic site. Several critical catalytic site amino acids (both polar and hydrophobic, designated as "SRS" or substrate recognition sites) interact closely with the docked carotenoid. The C4 carbon of the carotenoid is approximately 4 Angstroms above the activated iron-oxygen heme.
;){kind=link}
;){kind=link}
;){kind=link}
 β-Carotene Cleavage Dioxygenase. Carotene 15,15'-dioxygenase is a representative enzyme from this class, oxidizing the cleavage of β-carotene into two molecules of retinal (cleavage reaction). This enzyme has not been crystallized to date so the example shown on the right is Catechol 1,2-dioxygenase, a typical dioxygenase showing the single iron atom located at the catalytic site. Carotenoid cleavage enzymes are capable of oxidatively cleaving carotenoids at specific positions and have been identified in animals and plants. The first such enzyme to be identified was a 9-cis-epoxy carotenoid dioxygenase from maize, which catalyzes the rate-limiting step of abscisic acid biosynthesis. Similar enzymes are necessary for the synthesis of vitamin A in animals and other carotenoid-derived molecules in plants. In the model plant, Arabidopsis, there are nine hypothetical proteins that share some degree of sequence similarity to the 9-cis-epoxy carotenoid dioxygenases. Five of these proteins appear to be involved in abscisic acid biosynthesis. The remaining four proteins are expected to catalyze other carotenoid cleavage reactions and have been named carotenoid cleavage dioxygenases (CCDs). [Schwartz] CCDs have also been implicated in the cleavage of carotenoids in the formation of aromatic compounds - a fundmental process in flavor production in different plants and fruits. Plant volatiles function in a large number of ecological roles including attraction of insects for pollination and as initial indicators of plant suitability for herbivores. [Simkin]
β-Carotene Cleavage Dioxygenase. Carotene 15,15'-dioxygenase is a representative enzyme from this class, oxidizing the cleavage of β-carotene into two molecules of retinal (cleavage reaction). This enzyme has not been crystallized to date so the example shown on the right is Catechol 1,2-dioxygenase, a typical dioxygenase showing the single iron atom located at the catalytic site. Carotenoid cleavage enzymes are capable of oxidatively cleaving carotenoids at specific positions and have been identified in animals and plants. The first such enzyme to be identified was a 9-cis-epoxy carotenoid dioxygenase from maize, which catalyzes the rate-limiting step of abscisic acid biosynthesis. Similar enzymes are necessary for the synthesis of vitamin A in animals and other carotenoid-derived molecules in plants. In the model plant, Arabidopsis, there are nine hypothetical proteins that share some degree of sequence similarity to the 9-cis-epoxy carotenoid dioxygenases. Five of these proteins appear to be involved in abscisic acid biosynthesis. The remaining four proteins are expected to catalyze other carotenoid cleavage reactions and have been named carotenoid cleavage dioxygenases (CCDs). [Schwartz] CCDs have also been implicated in the cleavage of carotenoids in the formation of aromatic compounds - a fundmental process in flavor production in different plants and fruits. Plant volatiles function in a large number of ecological roles including attraction of insects for pollination and as initial indicators of plant suitability for herbivores. [Simkin]
References
1 - James Allen Olson and Norman I. Krinsky, Introduction: The colorful, fascinating world of the carotenoids: important physiologic modulators, FASEB 9:1547 (1995)
2 - George Britton, Structure and properties of carotenoids in relation to function, FASEB 9:1552 (1995)
3 - Hanyoup Kim et al., An Anomalous Distance Dependence of Intraprotein Chlorophyll-Carotenoid Triplet Energy Transfer, Biophysical Journal: Biophysical Letters, L28 (2005)
4 - Y. Nishida, Elucidation of a Carotenoid Biosyntheis Gene Cluster Encoiding a Novel Enzyme, 2,2'-β-Hydroxylase, from Brevundimonas sp. Applied and Environ. Microbiol. 71:8,4286-86 (2005)
5 - Garavelli, Marco et. al. DFT Study of the Reactions between Singlet-Oxygen and a Carotenoid Model, J. Am. Chem. Soc., 120(39), 10210-22 (1998)
6 - Thompson, A. J. et al., Plant Mol Biol. 2000 Apr;42(6):833-45
7 - Daisuke Umeno et al. Diversifying Carotenoid Biosynthetic Pathways by Directed Evolution, Microbiol. and Molec. Biology Reviews 69(1):51-78 (2005)
8 - George Britton, The biosynthesis of carotenoids: a progress report, Pure & Appl. Chem. 63(1): 101-8 (1991)
9 - Joonyul Kim et al., Defining the primary route for lutein synthesis in plants: The role of Arabidopsis carotenoid β-ring hydroxylase, PNAS 103(9): 3474-79 (2005)
10 - Li Tian et al., The Arabidopsis LUT1 locus encodes a member of the cytochrome p450 family that is required for carotenoid epsilon-ring hydroxylation activity, PNAS 101(1): 402-7 (2003)
11 - D. di Ricerca, Plant Carotenoids: functional genomic of xanthophylls biosynthesis and role in Arabidopsis thaliana, Universita degli Studi di Verona, Dept. Scientifico e Tecnologico (2005)
12 - Tomas Morosinotto et al. Mechanistic aspects of the xyanthophyll dynamics in higher plant thylakoids, Physiologia Plantarum 119:347-54 (2003)
13 - John Shanklin and Edgar B. Cahoon, Desaturation and Related Modifications of Fatty Acids, Annu. Rev. Plant Physiol. Plant Mol. Biol. 49:611?41 (1998)
14 - Efficient light harvesting through carotenoids. Thorsten Ritz, Ana Damjanovic, Klaus Schulten, Jian-Ping Zhang, and Yasushi Koyama. Photosynthesis Research, 66:125-144, 2000
15 - Energy transfer between carotenoids and bacteriochlorophylls in a light harvesting protein.
16 - Ana Damjanovic, Thorsten Ritz, and Klaus Schulten. Physical Review E, 59:3293-3311, 1999.
17- Fernando Fungo, et al., Correlation of fluorescence quenching in carotenoporphyrin dyads with the energy of intramolelcular change transfer states. Effect of the number of conjugated double bonds on the carotenoid moiety. Phys. Chem. Chem. Phys. 2003, 5, 469-475 (2003)
18 - Y. Demigiannakis, J. Hanley and A. W. Rutherford, J. Am. Chem. Soc., 2000, 122, 400?401.
19 - Chang-Lian Peng, et al. The antioxidative function of lutein: electron spin resonance studies and chemical detection, Functional Plant Biology, 2006, 33, 839-846.
20 - Hanyoup Kim et al., An Anomalous Distance Dependence of Intraportein Chlorophyll-Carotenoid Triplet Energy Transfer, Biophysical Journal: Biophysical Letters, L28 (2005)
21 - Steve's Place, academic notes on Photosynthesis designed for students and teachers.

Have a comment?
Be sure to drop by our Student/Teacher Biochemistry Weblog at http://www.3dbiochemblog.com.

This page was last edited on July 9, 2007