‘Ab initio’
Molecular Orbital Calculations
The electronic energy of a molecule can be calculated by
treating the overall electronic wavefunction as being a product of molecular
orbitals (strictly as a Slater determinant of molecular orbitals to satisfy the
Pauli principle; we should also include the electron spin).
So we can write a molecular wavefunction as:
Y = y1y2y3y4…yn (27)
Each molecular orbital, yi, is written as a
linear combination of atomic orbitals (the familiar LCAO approximation):
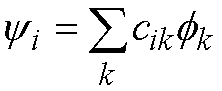 (28)
(28)
The molecular orbitals can be found by the Hartree-Fock
method. The Hamiltonian operator
contains terms for the potential and kinetic energy of the electrons. It includes the interactions between the
nuclei and the electrons, and the electron-electron repulsion. The molecular orbitals are found by making
an initial guess at the coefficients.
Then the determinant
½H–ES½= 0 (29)
H contains the
Hamiltonian matrix elements, and S
the overlap matrix elements. Solving
this determinant gives the molecular orbital energies E, which are substituted
into the secular equations to give new values of the coefficients ci. This whole process is repeated until the coefficients do not
change from one cycle to the next. The
orbitals are then described as self-consistent, and this process as a self-consistent
field procedure.
Each molecular orbital
is made up of a combination of atomic orbitals:
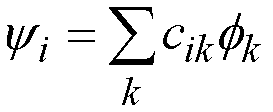 (28)
(28)
The set of atomic orbitals (fk) used is called
a basis set. When one function
is used for each filled atomic orbital (e.g. using 1s, 2s and 2p orbitals for a
carbon atom, this basis set is called a minimal
basis. Using more functions than
this will improve the calculation – it will increase the flexibility and
quality of the overall wavefunction.
From the variational principle we know that a better wavefunction will
give a lower energy. The bigger the basis
set, the better the calculation. If a
very large number of functions – a very large basis set – is used to represent
the atomic orbitals, we approach the Hartree-Fock limiting energy: the lowest
energy (best molecular wavefunction) that can be achieved by the Hartree-Fock
method.