Molecular Dynamics Simulations
· To begin,
require atomic coordinates and velocities:
· Generate
‘start-up’ configuration – all atoms assigned positions, ri,
and velocities vi.
· Velocities
chosen appropriate to a particular temperature (e.g. 300K) – see below
· Given potential
function V(r), calculate forces as:
F = -dV(r)/dr
Solve Newton’s
equations of motion:
Fi=miai
or d2ri/dt2 = Fi/mi
To obtain dynamic behaviour, need to solve 2nd order
differential equation for every particle in the system
ri(t+Dt) = ri(t) +viDt + 1/2ai(Dt)2
+ …
Typically, Leapfrog Verlet method used, with
timestep (Dt) approx. 1fs
(10-15s)
ri(t+Dt) = ri(t) +v(t–Dt/2) + mi–1Fi(t)Dt
vi(t+Dt/2) = vi(t –Dt/2) + mi–1Fi(t)Dt
So the positions of the atoms, and their velocities at a
susbsequent time t+Dt can be found.
This procedure is repeated thousands of times to allow equilibration, and
obtain an equilibrium distribution of velocities.
Unlike Monte Carlo, the temperature of the system is typically
not fixed. The temperature of the system can be calculated from the kinetic
energy of all the N atoms:
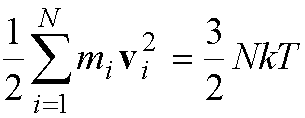
·
Special techniques may be needed to ensure that the temperature
does not change too far from the desired simulation temperature (e.g. by
scaling velocities).
·
Properties (e.g. thermodynamic averages) can be calculated from a
molecular dynamics simulation by a time average of the values for the
configurations generated in the simulation:
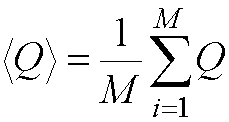
where M is the number of time
steps.
Molecular dynamics simulations are used for the same type of systems as Monte Carlo – they have been particularly useful for proteins and polymers, where it may be difficult to design good Monte Carlo moves. Molecular dynamics simulations are very important in the study of protein folding.
Comments
on the Monte Carlo and molecular dynamics simulation methods
· Obvious
difference: molecular dynamics provides information on time-dependence
of properties, Monte Carlo does not.
· Typical
conditions for dynamics simulation: constant number of particles (N),
constant volume (V), constant energy (E), the NVE
or ‘microcanonical’ ensemble.
· Typical
conditions for Monte Carlo simulation: constant number of particles (N),
constant volume (V) and constant temperature (T), the NVT or
‘canonical’ ensemble.
· MD has a
kinetic energy contribution, in MC the total energy is just calculated from the
potential function
· Monte Carlo
and molecular dynamics simulations are good for disordered systems, such melts,
liquids, solutions (in comparison with lattice dynamics which is good for
solids (ordered)).
· It is
difficult to model rare events (e.g. crossing barriers from one state to
another) as high energy configurations (e.g. transition states) are sampled
only rarely. Techniques such as ‘umbrella sampling’ are used to force a
system to change from one state into another (e.g. to force a chemical
reaction to occur in a QM/MM simulation). A ‘biasing’ potential forces the
system to move along a reaction coordinate to the transition state, and then to
the products.
· The
calculation of absolute free energies is very difficult with
either method. However, the convergence of the ensemble averages is very slow. Instead, relative
free energies can be calculated by gradually (and non-physically)
‘mutating’ one structure into another in a series of simulations, for example
in the free energy perturbation technique. One amino acid
sidechain could be mutated into another in a series of simulations to find the
effect on a protein.