Answers
for example questions for Molecular Modelling
Dr.
Adrian Mulholland
(1) What approximation leads to the concept of a potential energy
surface? What types of points on a
potential energy surface are particularly relevant in understanding a chemical
reaction?
The Born-Oppenheimer Approximation allows the separation of electronic
and nuclear motion, and so leads to the concept of a potential energy surface,
i.e. the electronic energy can be calculated at a particular fixed nuclear
geometry.
Two types of points are particularly relevant in understanding a
chemical reaction. Firstly, minima
correspond to stable molecular geometries (i.e. the structures of stable
molecules). First-order saddlepoints
are also useful. These points are an
energy maximum in one direction, but a minimum in all other directions. A first-order saddlepoint corresponds to a
transition state, i.e. the highest energy point along the reaction
pathway. The energy difference between
the minimum (corresponding to the reactants) and the transition state gives the
(potential) energy barrier to the reaction.
(2) Two methods which are widely used for the optimization of
molecular geometies are the ‘Steepest descents’ and ‘Newton-Raphson’
techniques. Without giving detailed mathematical
descriptions, briefly outline the advantages and disadvantages of these two
techniques.
The ‘steepest descents’ method of energy minimization is a simple and
effective method of improving the geometry of a molecular model (i.e. finding a
lower energy structure). It moves
‘straight downhill’ on the energy surface, taking steps in the direction of the
negative of the gradient. This involves
calculating the forces (gradients) on the atoms - the first derivative of the
energy). The gradient is a vector (g), with one component for each
coordinate. The step, s, is in the direction –g.
The steepest descents step is determined mostly by the largest
forces. It is therefore good for
relieving strain in a built model. Only
first derivatives are required - this is an advantage for large molecules. Steepest descents provides fast minimization
(big steps) when far from minimum, but small steps (slow minimization) when
close to minimum. It has slow
convergence (i.e. it will find the minimum energy structure only very
slowly). It may also oscillate around a
minimum and never actually reach the minimum.
Because it only moves ‘downhill’ on the energy surface, it is no use for
TS searches.
The Newton-Raphson method uses in addition second derivatives of the
energy and can be a better minimization method. It is efficient for small molecules, and converges quickly, in
contrast to steepest descents minimization.
However, it makes the approximation that the energy surface has a
quadratic form, which is often poor approximation for molecular potential
energy surfaces, particularly far from minimum The calculation, inversion and
storage of the Hessian matrix (i.e. the matrix of second derivatives) is
computationally difficult for large molecules.
It is possible to locate stationary points other than minima on
potential energy surfaces with the Newton-Raphson method, and this can be
useful for transition state optimization.
The Newton-Raphson method works well if the initial geometry is close to
a stationary point, but may not work well if it is not. Also, technically the method may be
difficult to use for some types of problem.
Technical difficulties arise because of the need to calculate the
Hessian, which is a potentially very large matrix (no. of variables
squared). The calculation of the second
derivatives can be demanding (requiring a lot of computer time), and their
storage can be difficult (a lot of computer memory is required). The Hessian must be inverted (diagonalized),
which is a technical problem for more than 1000 variables (1000 ´ 1000 matrix), i.e. approx.
300 atoms. It is possible to avoid
calculating the Hessian matrix at every step by either using a guessed matrix
(e.g. the identity matrix)or by using a Hessian update scheme (constructs an
approximate Hessian based on e.g. the gradient and coordinates at current and
new points. Calculating the Hessian at
a lower level of theory can also reduce the demands of the calculation. When dealing with large numbers of atoms,
though, the Newton-Raphson method is not suitable because of the large
computational requirements. A
first-order method has to be used instead (first-order methods use first
derivatives of the energy; second-order methods use first and second
derivatives).
(3) Give reasons why calculation and analysis of the harmonic
vibrational frequencies of molecules may be useful. Outline the process involved in calculation of harmonic
vibrational frequencies. In the case of
a diatomic molecule, find the frequency of vibration (treat the molecule as a
harmonic oscillator).
There are several reasons for calculating
vibrational frequencies of molecules, including:
· To identify the
nature of a stationary point found by geometry optimization (e.g. a minimum or
a saddlepoint). At a minimum, all the 3N-6
eigenvalues of the Hessian are positive, so all the calculated frequencies will
be positive.
· To calculate the
infrared and Raman spectra of a molecule
· To calculate
thermodynamic properties of a molecule
Vibrational frequencies are usually
calculated by finding the normal modes of the molecule.
for a harmonic oscillator, solving the
Schrödinger gives energy levels
Ev=(v+1/2)hn v=0,
1, 2….. (1)
This shows that an oscillator like this
cannot be at rest - the minimum vibrational energy it can have is hn/2 - the
zero-point energy.
The frequency, n, is related to
the force constant, k, for the
vibration:
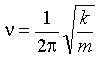 (2)
(2)
The force constant, k, is given by the second derivative of the potential energy, which
in one dimension is k=(d2V/dx2).
For a polyatomic molecule, the normal mode
frequencies are found from the matrix of second derivatives (the Hessian
matrix). It is important that the
second derivatives are mass-weighted - this is to take into account the fact
that a given force will different effects on atoms of different masses. For example, the zero-point energy is different
for a proton than for a deuteron in the same type of bond, due to the
difference in mass. Mass-weighting the
Hessian takes care of these effects.
The mass-weighted Cartesian coordinates (xm, ym, zm)
of any atom are related to its Cartesian coordinates (x,y,z) by:
xm = m1/2x ym = m1/2y zm = m1/2x (3)
where m
is the mass of the atom.
The mass-weighted Hessian matrix is given by:
F = M–1/2HM–1/2 (4)
M is a matrix containing the atomic
masses. Like the Hessian matrix, H, it
is a 3N ´ 3N matrix (N is the number
of atoms). M is a diagonal matrix - the
only non-zero elements are the masses.
Finding the eigenvalues (li) of the
mass-weighted Hessian matrix, F,
gives the frequencies of the normal modes, ni:
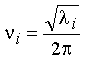 (5)
(5)
If Cartesian coordinates are used, there will
be 3N frequencies (from 3N eigenvalues), where N is the number of
atoms. Among these frequencies, 6 will
be very close to zero (5 for a linear molecule). These 6 correspond to overall translation and rotation of the
molecule. The remaining 3N-6 are vibrations of the molecule.
Example of a normal mode calculation: finding the
vibrational frequency of a diatomic molecule
The potential energy is taken here to be
harmonic,
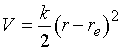 (6)
(6)
where r
is the separation of the two nuclei, and re
their equilibrium separation. We can
write this in terms of the Cartesian coordinates of the two atoms, x1 (for atom 1) and x2 (for atom 2) - we are
taking the bond to lie along the x
axis, and ignoring the y and z directions. Therefore, r = x2-x1. The second
derivatives of the potential energy are: d2V/dx12 = k, d2V/dx22 = k, d2V/dx1dx2 = d2V/dx2dx1 = –k.
Therefore the Hessian matrix (for the x direction only here) is:
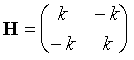 (7)
(7)
The mass-weighted matrix is:
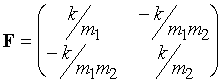 (8)
(8)
To find the eigenvalues, the secular
determinant is solved:
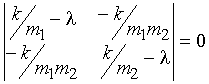 (9)
(9)
This gives a quadratic equation, with
solutions l1=0 and l2=k/m (where the
reduced mass m = m1m2/(m1 + m2)). The solution l = 0 corresponds
to a translation (in the x direction,
the only coordinate we are considering here).
The solution l = k/m gives the
frequency of the vibration - from equation (5) above,
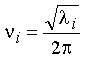
Therefore
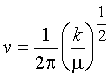 (10)
(10)
as expected for a harmonic oscillator.
In
general, calculations of vibrational frequencies (normal mode analyses) provide
the frequencies themselves (as the square roots of the eigenvalues), but also
the atomic displacements involved in each mode (from the eigenvectors), which
tell us what each separate vibrational motion looks like. Typically a calculation will print out the
frequency of each vibrational normal mode, and the associated atomic
displacements.
(4) Outline techniques which could be used to generate models of
transition state structures for chemical reactions. How could such transition state models be improved? Describe how you could confirm that a
structure really is a transition state structure. Also outline how you could show that the transition state really
is the right one for the reaction of interest.
For refinements of transition state
structures, it is essential to have starting models which are close to the real
transition state. Otherwise the
optimization will probably not be successful.
One way to generate a model of a transition state is to guess, using
chemical knowledge and intuition (e.g. identifying which bonds are breaking in
the TS, and so likely to be elongated).
However, this is difficult to do well for large systems.
A better approach in general is coordinate
driving (also called adiabatic mapping).
This involves optimizing the structure of the system with a particular
coordinate fixed. A sequence of
optimizations is performed with the coordinate fixed at a given value for each
optimization, then changed to a different fixed value for the next
optimization. This can generate an
energy profile, with the approximate TS at the highest point. For example, for ethane, optimizations could
be carried out with the value of one H-C-C-H torsion angle changed in a series
of steps from 60° to -60°. The energy
would be a maximum for the structure optimized with the H-C-C-H angle equal to
0°, which would be the model for the TS structure.
If the structures of the reactant and product
are known, can construct a model TS as a ‘midpoint’ between the two. For example, for the reaction H-N-C ® N-C-H, the TS model could
be specified from the internal coordinates for the reactant and product. The H-N-C angle in the reactant is 180°, whereas it is 0° in the
product. In the TS model, therefore, a
value of H-N-C = 90° could be used as a first guess. The bond lengths (interatomic distances) could also be chosen to
be intermediate between the reactant and product.
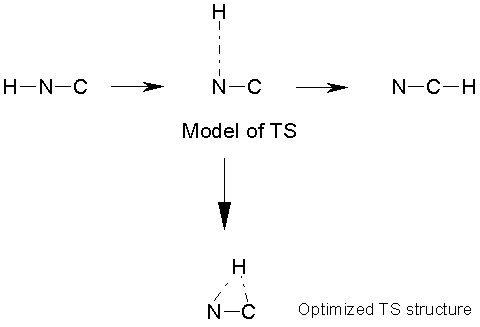
It is necessary to be careful in the choice
of coordinates- trying to create a model TS with Cartesian coordinates would
cause problems here (the hydrogen would end up between N and C!).
Alternatively, a TS model can be generated by
performing a geometry optimization with a particular symmetry enforced - the TS
may have a higher symmetry than the reactants or products. For example, the TS for the SN2
reaction Cl– +CH3Cl ® CH3Cl +Cl–
has D3h symmetry (the two
C-Cl distances will be the same, and the H atoms all lie in a plane):
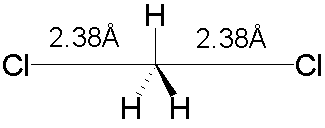
There is no single technique which can be
guaranteed to give a good TS model for all reactions. Finding a good model may be a matter of trial and error. A bad model will not optimize to the correct
TS.
To
check that the structure resulting from a geometry optimization really is a
transition state structure, a normal mode analysis (calculation of vibrational
frequencies) can be performed. For a transition state structure (saddlepoint),
one of the eigenvalues of the Hessian will be negative - i.e. the structure is
a maximum in one direction. The
frequency is given by the square root of the eigenvalue, so formally there will
be one imaginary frequency. If there is one, and only one, imaginary
frequency, the structure really is a transition state.
In
contrast, a minimum energy structure will have all positive frequencies (at a
minimum, all the 3N-6 eigenvalues of
the Hessian are positive, so all the calculated frequencies will be positive).
If there is more than one imaginary frequency, the structure is a higher order
saddlepoint (a maximum in more than one direction), and chemically not very
interesting - you would need to do more geometry optimization to find a real TS
structure.
It is still
possible, however, that the TS structure found by a geometry optimization is
the TS for a process different to the one of interest. To check that the TS structure really is the
desired TS, examining the atomic displacements for the reaction coordinate can
show which atoms are involved (the atomic displacements for the imaginary
frequency are very important - this normal mode corresponds to motion along the
reaction coordinate). To prove
that it really is the desired TS, calculating the steepest descents path from
the TS should produce (in one direction) the reactants and (and in the other
direction) the products. This path
calculated in mass-weighted coordinates from the TS is called the intrinsic
reaction coordinate.
(5) Find the Newton-Raphson step, and the minimum, for the
function f(x,y) = 2x2 + 2y2, starting from the point (x,y) = (3,–2). Show your working.
The Newton-Raphson expression for stepping towards a minimum for a
multidimensional function is
xk+1 = xk – gH–1
For a molecule with N atoms, there are 3N
Cartesian coordinates, so the gradient is a vector g (as above), with 3N
terms (dV/dxi).
The second derivatives have
the form (d2V/dxidxj), involving each
coordinate, and so make up a 3N´3N matrix, called the Hessian matrix, H.
The energy of a particular molecule can be given as a function of two
coordinates:
E(x,y)
= 2x2 + 2y2
Find the Hessian and inverse Hessian matrices for this system.
The Hessian, the matrix of second derivatives
of the function, i.e. for this 2-dimensional function, a 2´2 matrix:
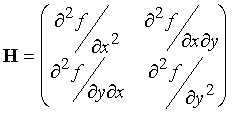
For f(x,y)=x2
+ 2y2, d2f/dx2 = 2; d2f/dy2 = 4; d2f/dxdy = 0, so:
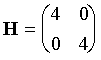
and the inverse matrix is
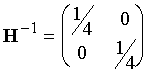
Give the value of the Newton-Raphson step for (x,y) = (3,–2) and give
the predicted coordinates of the minimum of the function.
The gradient is (df/dx = 4x ; df/dy = 4y):
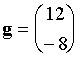 for (x,y)=
(3,–2)
for (x,y)=
(3,–2)
So the new coordinates are

The minimum is the point x=0, y=0. For a purely quadratic function like
this one, the Newton-Raphson method finds the minimum in a single step from any
point on the surface.
(6) Briefly describe the following methods for calculating
molecular energies and geometries (outline their advantages and disadvantages):
(i)
ab initio molecular orbital methods
(ii)
semiempirical molecular orbital methods
(iii)
molecular mechanics methods
(iv)
combined quantum mechanics/molecular mechanics methods
(i) Ab initio molecular orbital calculations can provide accurate
geometries and energies, if a high enough level of theory is used. In many cases, using a large basis set and a
method which includes electron correlation can provide good answers. However, such calculations are extremely
demanding of computational resources, and so are feasible only for small
molecules. The most basic form of ab
initio calculations use the Hartree-Fock method. These can be performed reasonably quickly, even on fairly large
molecules (with e.g. 10-100 heavy atoms).
Hartree-Fock calculations can be useful for determining geometries of
stable molecules. The best results are
achieved with large basis sets.
However, they do not include electron correlation, and so they do not
provide a good description of chemical reactions (they deal poorly with
situations where bonds are partially formed or broken).
(ii)
Semiempirical Methods Ab initio
calculations require a lot of computer time and memory. This is because many difficult integrals
involving the atomic orbitals have to be calculated for the electron-electron
interactions. So-called ‘semiempirical
methods’ are much faster, and can be applied to bigger molecules (e.g. 100s of
atoms instead of 10s of atoms). In
semiempirical methods, fewer integrals have to be calculated because
simplifying approximations are made.
The integrals that do remain are not calculated directly, but instead
are replaced by simple functions. These
simple functions are parameterized so that the semiempirical method reproduces
experimental data (e.g. molecular structures, dipole moments, ionization
energies, etc.) as well as possible.
The experimental data contain the effects of electron correlation, and
so semiempirical methods can be a good choice in some cases.
However, the approximations
used make them often somewhat inaccurate.
Also, they can only reproduce properties of molecules similar to those
used in parameterizing the method. Usually
only data for stable molecules are included, which means that transition states
may not be dealt with well. Examples of
semiempirical molecular orbital methods include MNDO, AM1 and PM3.
(iii) Molecular mechanics.
Even faster (‘cheaper’) calculations of the structures and energies of
molecules are possible using ‘molecular mechanics’. These methods forget about electrons and nuclei, and treat atoms
as point charges with van der Waals radii.
Bonds are treated as springs (simple harmonic energy terms), with simple
terms also for torsion angles and bond angles.
These functions contain parameters (bond lengths, force constants, etc.)
which are optimized to reproduce the structures of molecules as found experimentally
or by ab initio calculations.
EMM = Ebond + Eangle + Etorsion
+ EVDW + Eelectrostatic
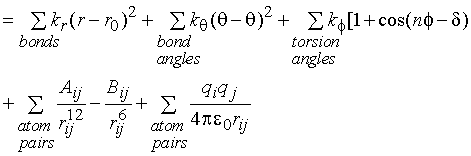
With molecular
mechanics methods, it is possible to study very large molecules (solids,
polymers, surfaces, proteins, DNA, etc.).
The motion of these molecules can be studied using molecular mechanics
energy functions in molecular dynamics simulations. However, molecular mechanics potential functions are not suitable
for modeling chemical reactions. The
functional form is incorrect (e.g. a harmonic term for stretching a bond does
not allow the bond to break); also the use of stable molecules in the
parameterization process means that only stable molecules can be described by
these functions, and not e.g. transition states.
(iv) Combined quantum
mechanical/molecular mechanical (QM/MM) methods. To treat a chemical reaction, a quantum mechanical method is
required to describe the breaking and forming of bonds - such methods include
ab initio and semiempriical molecular orbital methods. Molecular mechanics methods are not suitable,
as outlined above. However, ab initio
methods cannot be applied to systems with more than a few tens of atoms,
because the computational demands are too great. Semiempirical methods can be applied to bigger systems (100s of
atoms), but even they are too demanding for calculations on very large
systems. For calculations on reactions
in solution, where thousands of solvent molecules may need to be included, or
for reactions in solids, at surfaces or in biological macromolecules (e.g.
enzymes), a different approach must be used.
Quantum mechanical/molecular
mechanical (QM/MM) methods allow reactions in large systems to be studied. A small region including the reacting atoms
is treated with a QM method (e.g. ab initio molecular orbital theory), while
most of the system is represented more simply by molecular mechanics. The reacting region ‘feels’ the influence of
the molecular mechanics environment - for example, the atomic charges of the MM
atoms affect the QM atoms. By combining
the accuracy of a QM method (for a small reacting system) with the speed of a
MM method (for its surroundings), reactions in large molecules and in solution
can be studied.

Schematic
representation of the setup of a QM/MM calculation. A small region containing the reacting atoms is treated by a
quantum mechanical method (e.g. by molecular orbital theory), while its
surroundings are represented more simply by molecular mechanics.
(7) What is meant by the term ‘correlation energy’? What methods can be used to calculate
energies including correlation effects?
Correlation
energy
The Hartree-Fock
method has important limitations. Even
the Hartree-Fock limiting energy is above the true energy of a molecule. This is because correlation energy is not
included. The correlation energy can be
defined as:
Ecorrelation = Etrue – EHartree-Fock (30)
This error arises because the interaction of one electron with another
is treated as the interaction with a smoothed out, averaged electron
density. In fact, the position of one
electron affects the position of the other electron, because they repel one
another. If one is on one side of the
molecule, the other electron is likely to be on the other side. Their positions are correlated, an effect
not included in a Hartree-Fock calculation.
While the absolute
energies calculated by the Hartree-Fock method are too high, relative energies
may still be useful. If the correlation
energy does not change during a process, the energy calculated for the process
with the Hartree-Fock method will be close to the real value. The correlation energy does not change very
much if the bonding in the molecule does not change much. So, for example, calculations of
conformational properties can be done accurately at the Hartree-Fock level
(e.g. calculating the rotational barrier in ethane). When the bonding in a molecule changes, though, the correlation
energy changes significantly. This
means that the Hartree-Fock method performs very poorly for calculations on
breaking chemical bonds. The
dissociation energy predicted is too high, and it may predict the wrong
products to be formed (e.g. predicting the formation of ions when in fact
neutral atoms should be formed by breaking a bond).
For treating chemical
reactions, we need to use methods which include electron correlation. Many methods of this type use a Hartree-Fock
calculation as a starting point. An
example is the use of perturbation theory, developed by Møller and
Plesset and available in many ab initio molecular orbital programs. The term MP2 is used for second-order
perturbation theory (the results given above for the Diels-Alder, SN2
and Claisen rearrangement reactions were calculated at the MP2 level). A more sophisticated (and computationally
demanding) approach is called configuration interaction (CI). In this technique, different electronic
configurations are allowed to interact, to give a better overall
wavefunction. The improved,
configuration interaction, molecular wavefunction is written as a linear combination:
Yimproved, CI = aY0 + bY1 + cY2 + ….
Y0 is the wavefunction found first (e.g. from a
Hartree-Fock calculation). Y1, Y2 etc. are
wavefunctions based on the original Y0 in which some electrons have
been moved into higher energy molecular orbitals which are unoccupied in Y0. This gives a better overall wavefunction, Yimproved, CI. CI calculations tend to be extremely
demanding of computer resources, because many excited electronic configurations
(Y1, Y2…) are
possible. Usually only a limited number
can be included.
Recently, techniques
based on density-functional theory have become popular. The energy of the molecule is calculated as
a function (strictly a functional) of the electron density. Density-functional theory calculations on
molecules usually include a Hartree-Fock calculation. They are popular because they include electron correlation, but
are not as demanding of computer resources as methods such as MP2 and CI.
(8) The Claisen rearrangement of allyl vinyl ether to form
5-hexenal is found to have an activation enthalpy, DH‡, of 92.5 kJ mol-1 and an activation
entropy DS‡
of –0.038 kJ mol-1
K-1. Calculate the
first-order rate constant of this reaction at 300K.
At 300K, the activation free energy for the reaction is given by:
D‡G = D‡H – TD‡S
D‡G = 92.5 – (300 ´ –0.038)
D‡G = 92.5 + 11.4 =
103.9 kJ mol-1
A (first-order) rate constant can be calculated from
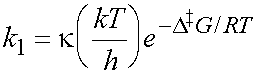
k is the transmission coefficient. For most reactions, k is close to 1,
so we can put k = 1 here (use this approximation generally, unless
a specific, different value of k is given).
(kT/h) = (1.380 ´ 10-23
´ 300) / (6.626 ´ 10-34) = 6.2 ´ 1012 s-1
exp (–103,900/ (8.315 ´ 300) = exp
–41.65 = 8.159 ´ 10-19
(be careful to use Joules (not kJ) for D‡G, as R = 8.134 J K-1 mol-1).
So the rate constant for the reaction is 8.159 ´ 10-19 ´ 6.2 ´ 1012 s-1 = 5.1 ´ 10-6 s-1
(9) The enzyme citrate synthase catalyses the formation of
citrate, the first step in the central metabolic pathway, the citric acid (or Krebs)
cycle. The enzyme is found to have a
maximum turnover number (see e.g. Atkins, ‘The Elements of Physical Chemistry,
(3rd ed.) pg. 252) of 100s-1 at 298K. What is the activation free energy for the enzyme-catalysed
reaction at this temperature?
The maximum turnover number can be treated as an effective first-order
rate constant (from the Michaelis-Menten mechanism for enzyme action).
It can therefore be expressed as
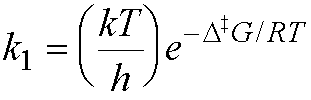
Therefore, the activation free energy is given by:
D‡G =
RT ln
(hk1/kT)
= – 8.314 ´ 298 ln (100/6.2 ´ 1012)
= - 8.314 ´ 298 ´ -24.85
= 61569 J mol-1
= 61.6 kJ mol-1
Exam question from 2003:
Answer part (a) or part (b)
(a)
Give an expression for a ‘molecular mechanics’ potential energy
function of the type used for standard macromolecular simulations. Include
intra- and intermolecular terms. State clearly what each energy term
represents, and its physical origin. How, typically, would the parameters of
such a potential energy function be found? Discuss the range of applicability
of a potential energy function of this type, its strengths and its limitations.
Give brief examples of applications. What improvements could be made to a
potential energy function of this type?
The molecular mechanics potential functions discussed in the course
have the general form:
EMM = Ebond + Eangle + Etorsion
+ EVDW + Eelectrostatic
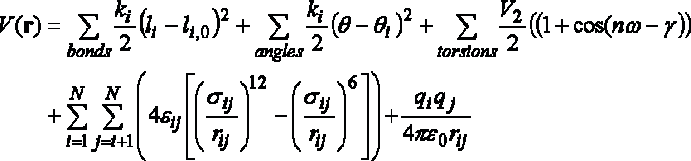
The answer should discuss the harmonic function used to model bond
stretching, why this is chosen over e.g. a Morse potential, the limitations of
the harmonic potential (e.g. no dissociation; poor behaviour away from
minimum); the harmonic valence angle bending; the cosine series for torsion
angles, mentioning in each case the meaning of the symbols (i.e. force
constants, barrier heights, phase factors). The possibility of bonded cross
terms (e.g. stretch-bend coupling) should be outlined, together with possible
functional forms (e.g. Urey-Bradley 1-3 terms). The final terms should be
identified as the non-bonded interactions in the potential function, van der
Waals and electrostatic terms. The answer should discuss which non-bonded
interactions would be calculated (i.e. 1-4 interactions etc). In discussing the
van der Waals terms, the repulsive and attractive interactions should be
identified, and the cause (dispersion interactions and exchange repulsion)
should be outlined. Atomic partial charges should be described, with typical
values (e.g. relating to electronegativity), and the ability of these models to
reproduce hydrogen bond structures. Limitations of molecular mechanics force
fields such as lack of electronic polarizability (fixed atomic charges),
inability to model reactions (incorrect functional form), dependence on
parameterization (e.g. limited dataset), should be discussed. Practical improvements
include: inclusion of atomic polarizability (simple models should be given),
different forms for bonding interactions; and combined quantum
mechanics/molecular mechanics (outline briefly) - allow modelling of reactions.
(b) Discuss the Monte Carlo and molecular dynamics simulation methods.
Outline the techniques by which each is carried out. Compare and contrast the
two techniques. Briefly also discuss the lattice dynamics technique.
Describe basic features of MD and MC. Outline integration algorithms
for MD, typical timesteps, use of SHAKE algorithm for increasing timestep. For
MC, outline importance sampling, Metropolis method, random number generation,
Monte Carlo 'moves'. Sampling efficiency. Classical MD determines atomic
positions through solving Newton's equations of motion. Differences between MC
and MD: e.g. MD gives information on time dependence of properties, MC does
not. MD has a kinetic energy contribution to the total energy, whereas MC only
includes potential energy. Standard MD would be performed in the NVE ensemble,
MC would be performed in canonical ensemble (N,V,T) (although both methods can
be modified to sample from other ensembles). Definition of temperature in MD.
Lattice dynamics - brief outline of methods. Calculation of free energies
through lattice dynamics - normal mode analysis, calculation of free energy,
enthalpy and entropy through consideration of harmonic vibrational frequencies
and partition functions.