Computational chemistry and molecular modelling
Approximate molecular orbital methods, such as Hückel Theory, have been useful in helping chemists develop ideas and understanding of molecules.
However, the
development of computers, and sophisticated ‘ab initio’ molecular orbital
methods (based on the same basic approaches such as the variational principle),
mean that it is now possible to calculate molecular properties on desktop
PCs. Nowadays, computers are important
tools in nearly all areas of chemical research. For example:
· Pharmaceutical
industry (e.g. drug design)
· Materials science
· Biotechnology
· Catalysis (e.g
modelling catalysis in zeolites and transition metal complexes)
· Spectroscopy
· Atmospheric
chemistry
· Crystallography
· Reaction dynamics
as well as
investigating organic reactivity problems.
Modelling and
calculations can given insight beyond experiments in many cases. Modelling and
experiments are best done in conjunction: modelling helps interpret experiments, and
experiments test computer models.
Many different computational chemistry and molecular modelling methods are used - you’ll learn about some of these in the future.
To understand a chemical reaction, we need to know the transition state structure for the reaction. Transition states are difficult to study directly by experiment. Instead, their structures can be calculated and analysed.
Modelling
reactivity: calculations can make useful predictions even for reactions in
large, complex systems like enzymes
Calculated
transition state structure for the reaction in the enzyme phenol hydroxylase
(an OH group is transferred to the ortho
carbon of phenol):
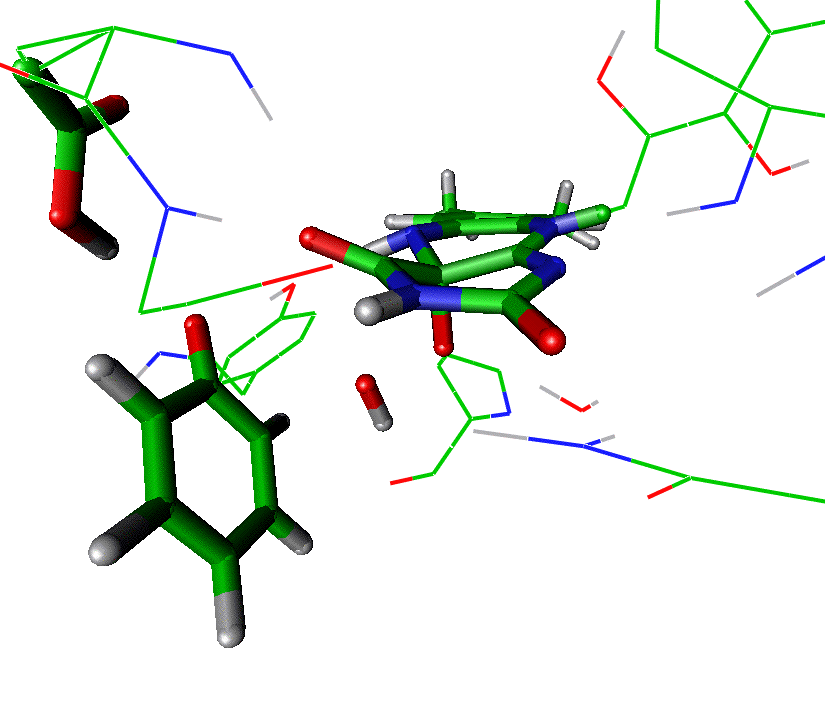
(Phenol
hydroxylase is involved in biodegradation of aromatic pollutants).
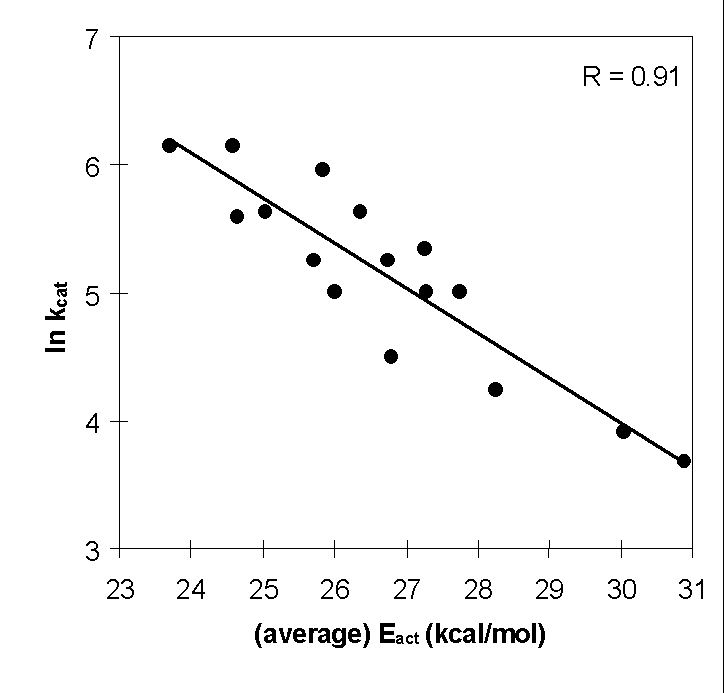
Linear correlation between the natural logarithm of
the experimental rate constants and the calculated energy barriers (Eact)
for conversion of phenol derivatives by phenol hydroxylase