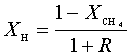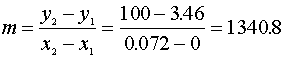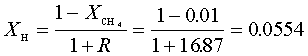Chapter 8 - CHEMKIN Simulations
8.1
Introduction
In-situ molecular beam mass spectrometry
(MBMS) of diamond CVD environments has proved both useful and versatile in its
application. However, detailed
numerical modelling of the CVD gaseous environment can serve not only as a
useful complement to experimental studies but also as an important instrument
to test the validity of the MBMS results.
To develop such a modelling system is not trivial by any means since
chemical reaction mechanisms containing hundreds of reactions and involving 50
or more chemical species are common in the diamond CVD process. Thus there is a need for accurate, detailed
descriptions of the chemical kinetics occurring in the gas-phase and/or on
reactive surfaces, in order to perform a complete simulation of the diamond
growth process.
In
the present study, a highly structured computer package called CHEMKIN8.1
has been employed to aid in the incorporation of a complex gas-phase chemical
reaction mechanism into numerical simulations.
Such a package has already been utilised by several groups8.2-8.7
as a means of characterising the diamond growth environment using different
source gas mixtures. Particularly
beneficial to the present work is the capacity to calculate concentrations of
free radicals as well as stable species, provided the necessary input data
relevant to the species is given to complement the measured concentrations of
the stable species.
8.2
Structure of CHEMKIN
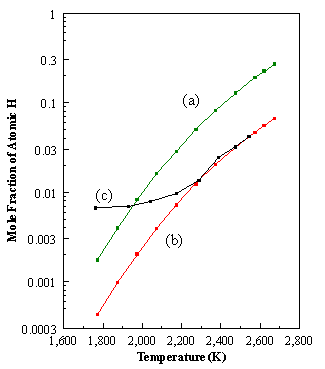
CHEMKIN is a highly structured package that requires the manipulation of a number of programs, subroutines, and data files. Figure 8.1 shows a schematic diagram of the general structure of the CHEMKIN package. It is composed of four important pieces: the Interpreter, the Thermodynamic Database, the Linking File and the Gas-Phase Subroutine Library.
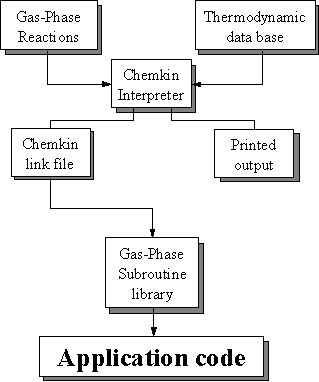
Figure 8.1. Structure of the CHEMKIN package and its link to an application
code (adapted from Reference 8.1).
The Interpreter is a program
that first reads the user’s symbolic description of the reaction mechanism,
where the pre-exponential factor Ai
(units - cm-mol-s-K) , the temperature exponent bi, and the activation energy Ei
(units - cal/mol) are specified (See Figure 8.2).
Consider the rate law for the consumption of reactant A as:
![]()
where k
is the rate constant, and the reaction is second order overall, but first order
in each of two reactants A and B. The
rate constant, k, is related to the
pre-exponential factor A, the
temperature exponent b, and the activation energy E via
![]()
In the above rate
law k will have units:
![]()
k = cm3mol-1s-1
and so the units of the pre-exponential
factor, A, will be written as cm3mol-1s-1. However, we parametise the traditional A
factor to include a temperature-dependent term, i.e. AiTb. This term will therefore have units cm3 mol-1s-1. So if b = 1, for example, then Ai will have units mol-1cm3s-1K-1. Clearly if b is non-integer then the
units of Ai will be mol-1cm3s-1K-b.
Having read the symbolic
description of the reaction mechanism the CHEMKIN package then extracts
thermodynamic information for the relevant species from the Thermodynamic Data
Base.8.8 The information in
the database can be added or modified by input to the Interpreter (See
Figure 8.3). The thermodynamic data
are stored as polynomial fits to specific heat cp/R, enthalpy
H0/RT, and entropy S0/R.
There are seven coefficients for each of two temperature ranges.
![]() (1)
(1)
![]() (2)
(2)
![]() (3)
(3)
Thus for each species there are 14 coefficients in all. In addition to the polynomial coefficients for each species, the Thermodynamic Data Base provides the species name, its elemental make-up, and the temperature ranges over which the fits are valid.
Essentially the thermodynamic database is a table of specific heat, enthalpy, and entropy as a function of temperature. In addition, since the fits span two temperature ranges, the temperature ranges have to be specified. A common temperature is used which connects the two ranges, typically 1000K, but it may be different in some cases. An illustration of the thermodynamic data format is shown below.
! CHEM.INP Figure 8.2. The input file used by the
! Input file for CHEMKIN interpreter. Interpreter in this present study. The
ELEMENTS gas
phase reactions was obtained
C H from Refs. 8.9, 8.10 and 8.11
END
SPECIES
CH4 CH3 CH2 CH C
C2H6 C2H5 C2H4 C2H3 C2H2 C2H
H H2
END
REACTIONS
! ---------------------------------HYDROGEN
REACTIONS------------------------------
H+H+M=H2+M 9.7E+16 -0.6
0
! --------------------------------------C-1
REACTIONS--------------------------------------
CH3+H+M=CH4+M 8.0E+26 -3.0
0
CH4+H=CH3+H2 2.2E+04 3.0 8800
CH4+CH2=CH3+CH3 1.0E+13 0.0
0
CH3+CH3=C2H5+H 2.8E+13 0.0 13600
CH3+CH3+M=C2H6+M 3.2E+41 -7.0
2766
CH3+H=CH2+H2 7.2E+14 0.0 15100
CH3+CH2=C2H4+H 2.0E+13 0.0
0
CH3+M=CH2+H+M 1.0E+16 0.0 90600
CH2+H=CH+H2 4.0E+13 0.0
0
CH+CH4=C2H4+H 6.0E+13 0.0
0
CH+CH3=C2H3+H 3.0E+13 0.0
0
CH+H=C+H2 1.5E+14 0.0
0 !C+F 92:145-160
CH+CH2=C2H2+H 4.0E+13 0.0
0 !C+F 92:145-160
C+CH3=C2H2+H 5.0E+13 0.0
0 !C+F 92:145-160
C+CH2=C2H+H 5.0E+13 0.0
0 !C+F 92:145-160
! -------------------------------------C-2
REACTIONS--------------------------------------
C2H6=CH3+CH3 9.4E+45 -9.0
100700
C2H6+H=C2H5+H2 5.4E+02 3.5
5210 !C+F 92:419-439
C2H6+CH3=C2H5+CH4 5.5E-01 4.0
8300
C2H5+M=C2H4+H+M 1.0E+17 0.0 31000
C2H5+CH3=C2H4+CH4 7.9E+11 0.0
0
C2H4+M=C2H2+H2+M 2.6E+17 0.0 79400
C2H4+M=C2H3+H+M 2.6E+17 0.0 96600
C2H4+H=C2H3+H2 1.1E+14 0.0
8500 !C+F 92:419-439
C2H4+CH3=CH4+C2H3 4.2E+11 0.0 11200
C2H3+H=C2H2+H2 2.0E+13 0.0
0
C2H3+M=C2H2+H+M 3.0E+15 0.0 32000
C2H3+CH3=C2H2+CH4 7.9E+11 0.0
0
C2H2+M=C2H+H+M 4.0E+16 0.0 107000
C2H2+H=C2H+H2 6.0E+13 0.0 23700
C2H2+CH3=C2H3+CH2 3.1E+14 0.0 69888 !C+F
92:419-439
C2H2+C2H2=C2H3+C2H 3.1E+14 0.0 91208 !C+F
92:419-439
C2H2+CH2=C2H3+CH 1.5E+14 0.0 62636 !C+F
92:419-439
C2H2+H2=CH2+CH2 7.1E+15 0.0 127788 !C+F 92:419-439
END
|
Name of species CH4 |
Date ¬ atomic phase symbols of & species formula ® 121286C 1H 4 G |
low high common temp. temp. temp. (K) (K) (K) 0300.00 5000.00 1000.00 |
Line number 1 |
||
|
a1 0.01683478E+02 |
a2 0.10237236E-01 |
a3 -0.03875128E-04 |
a4 0.06785585E-08 |
a5 -0.04503423E-12 |
2 |
|
a6 -0.10080787E+05 |
a7 0.09623395E+02 |
a1 0.07787415E+01 |
a2 0.01747668E+00 |
a3 -0.02783409E-03 |
3 |
|
a4 0.03049708E-06 |
a5 -0.12239307E-10 |
a6 -0.09825229E+05 |
a7 0.13722195E+02 |
|
4 |
Figure 8.3. Format for thermodynamic data. The first
seven coefficients in the polynomial expansion are for the upper temperature interval. The final seven
coefficients in the polynomial expansion are for the lower temperature interval.
In addition to printed output, the Interpreter writes a Linking File, which contains all the pertinent information on the elements, species, and reactions in the mechanism. Once the Interpreter has been executed and the Linking File created, the Gas-Phase Subroutine Library can be used. These subroutines are called from the Fortran code. The CHEMKIN gas-phase subroutines, of which there are over 100, are called as required to return information on the elements, species, reactions, equations of state, thermodynamic properties and chemical production rates. In general the input to these routines is the state of the gas-pressure or density, temperature, and the species composition. The code is designed to facilitate selection of the particular CHEMKIN subroutines that are needed for a given problem.
8.3
Structure of the Transport Property Fitting Code and SURFACE CHEMKIN
Since
the diamond CVD process not only involves gas-phase reactions mechanism but
also heterogeneous gas-solid reactions occurring on the substrate and the
various transport processes, the CHEMKIN package alone cannot fully simulate
the growth process. The Transport
package and SURFACE CHEMKIN8.12 are therefore introduced as another
component of a large body of software designed to facilitate the computational
modelling of chemical kinetics in flowing systems such as diamond CVD. Figure 8.4 shows how these packages are
linked and used in the present study to simulate the diamond CVD process.
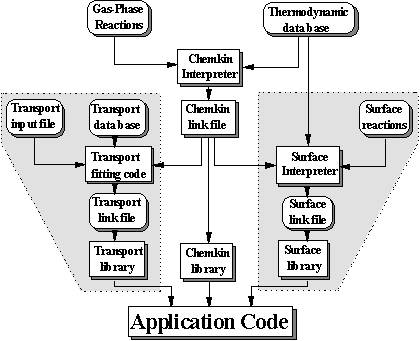
Figure 8.4. Relationships and flow of information
between the CHEMKIN, Transport, and SURFACE CHEMKIN packages, and a user’s
application code (adapted from Reference 8.12).
The Transport package handles the gas-phase molecular transport properties and SURFACE CHEMKIN handles the surface thermodynamics and chemical kinetics. Each package consists of an Interpreter, a database of either thermodynamic or transport properties, and a library of subroutines that can be called from the application code.
In the case of diamond CVD, since the surface chemistry involves gas-solid heterogeneous reactions, the first step is to run the CHEMKIN Interpreter, which reads the user’s description of the gas-phase reaction mechanism (Figure 8.2). The Linking file is read by the Gas-Phase Subroutine Library that makes the information available to all the other subroutines in the library.
The next step is to execute the SURFACE CHEMKIN Interpreter, which reads the user’s symbolic description of the surface reaction mechanism. Required thermodynamic data can come from the same Thermodynamic Database used by CHEMKIN or from a separate Thermodynamic Database compiled for surface reactions. As Figure 8.5 shows, the thermodynamic data have been added by input into the Interpreter. The SURFACE CHEMKIN Interpreter extracts all needed information about gas-phase species from the CHEMKIN Linking File. Like the CHEMKIN Interpreter, the SURFACE CHEMKIN Interpreter also provides a printed output and a Linking File. Again the Surface Linking File is read by a subroutine in the Surface Subroutine Library that makes the surface reaction mechanism information available to all other subroutines in the library.
Finally, the Transport Property Fitting Code reads the CHEMKIN Linking File and identifies all the gas-phase species that are present in the gas-phase reaction mechanism. Then, drawing on a database of molecular parameters, it computes the species’ viscosities, thermal conductivities, and diffusion coefficients. As with the other packages it provides a Linking File that is read by a subroutine in the Transport Property Subroutine Library. Subroutines from this library may be called by the application code to return transport properties for individual species or for a multicomponent gas mixture.
! SURF.INP
! Input file for SURFACE interpreter.
!
! SURFACE SITES & SPECIES.
! Density of 5.22E-09 mol/cm2 corresponding to sites on the (111) surface of diamond.
!
SITE/DIAMOND/ SDEN/5.22E-09/
CH(S) C(S,R) C(R,G) C(S,R3) HCCH(S,R)
CH2(S) CH(S,R) CH(S,G) CH(S,R2) H2CCH(S)
CH3(S) CH2(S,R) CH2*(S)
END
!
BULK
D /3.515/
G /3.515/
END
!
! THERMODYNAMIC DATA.
! Taken from Tables III and IV of Reference 8.13
!
THERMO ALL
300. 1000. 3000.
CH(S) 050595C 1H 1 0 0G 300.00 3000.00 1000.00 1
0.14872259E+01 0.33000924E-02 -0.28411702E-06 -0.34383971E-09 0.76660243E-13 2
-0.05865759E+04 -0.09239365E+02 0.14872259E+01 0.33000924E-02 -0.28411702E-06 3
-0.34383971E-09 0.76660243E-13 -0.05865759E+04 -0.09239365E+02 4
C(S,R) 050595C 1 0 0 0G 300.00 3000.00 1000.00 1
0.16900997E+01 0.11069085E-02 -0.12616481E-06 0.11996654E-09 0.28811839E-13 2
0.02130371E+06 -0.09750481E+02 0.16900997E+01 0.11069085E-02 -0.12616481E-06 3
-0.11996654E-09 0.28811839E-13 0.02130371E+06 -0.09750481E+02 4
C(S,R3) 100191C 1 0 0 0G 300.00 3000.00 1000.00 1
0.16900997E+01 0.11069085E-02 -0.12616481E-06 -0.11996654E-09 0.28811839E-13 2
0.64193504E+05 -0.10340458E+02 -0.12628431E+01 0.73135700E-02 -0.72650602E-06 3
-0.57274971E-08 0.29829661E-11 0.65054285E+05 0.52818985E+01 4
CH(S,R2) 062992C 1H 1 I 300.00 3000.00 1000.00 1
0.14872311E+01 0.33000871E-02 -0.28411483E-06 -0.34383973E-09 0.76660114E-13 2
0.43930898E+05 -0.10147053E+02 -0.18659885E+01 0.90232668E-02 0.29341194E-06 3
-0.52555555E-08 0.20408896E-11 0.45065105E+05 0.81504097E+01 4
CH(S,G) 050792C 1H 1 I 300.00 3000.00 1000.00 1
0.14817694E+01 0.32751665E-02 -0.27781763E-06 -0.34004749E-09 0.75557168E-13 2
0.10660619E+05 -0.10097240E+02 -0.18745055E+01 0.90725571E-02 0.24365704E-06 3
-0.53500671E-08 0.21229865E-11 0.11787394E+05 0.81871119E+01 4
HCCH(S,R) 071291C 2H 2 0 0G 300.000 3000.000 1000.00 1
0.17394471E+01 0.51764320E-02 -0.42153641E-06 -0.53463645E-09 0.11763388E-12 2
-0.59624932E+04 -0.12329198E+02 -0.25071146E+01 0.12199585E-01 0.59056043E-06 3
-0.66420434E-08 0.24363521E-11 -0.45017656E+04 0.10931940E+02 4
CH2(S) 050595C 1H 2 0 0G 300.00 3000.00 1000.00 1
0.17394471E+01 0.51764320E-02 -0.42153641E-06 -0.53463645E-09 0.11763388E-12 2
-0.06534599E+05 -0.11177380E+02 0.17394471E+01 0.51764320E-02 -0.42153641E-06 3
-0.53463645E-09 0.11763388E-12 -0.06534599E+05 -0.11177380E+02 4
CH(S,R) 050595C 1H 1 0 0G 300.00 3000.00 1000.00 1
0.14872259E+01 0.33000924E-02 -0.28411702E-06 -0.34383971E-09 0.76660243E-13 2
0.01547748E+06 -0.09239365E+02 0.14872259E+01 0.33000924E-02 -0.28411702E-06 3
-0.34383971E-09 0.76660243E-13 0.01547748E+06 -0.09239365E+02 4
C(R,G) 050792C 1 I 300.00 3000.00 1000.00 1
0.16900998E+01 0.11069081E-02 -0.12616400E-06 -0.11996697E-09 0.28811900E-13 2
0.35690953E+05 -0.10340459E+02 -0.12628419E+01 0.73135681E-02 -0.72650272E-06 3
-0.57275016E-08 0.29829680E-11 0.36551734E+05 0.52818918E+01 4
H2CCH(S) 071291C 2H 3 0 0G 300.00 3000.00 1000.00 1
0.36940942E+01 0.79702111E-02 -0.64374024E-06 -0.81818519E-09 0.17982379E-12 2
0.14525338E+05 -0.24731092E+02 -0.43835902E+01 0.23068689E-01 -0.28454056E-0 3
-0.14521289E-07 0.65029358E-11 0.17095203E+05 0.18776432E+02 4
CH3(S) 050595C 1H 3 0 0G 300.00 3000.00 1000.00 1
0.22271934E+01 0.64840489E-02 -0.50900690E-06 -0.66263206E-09 0.14445464E-12 2
0.07774954E+05 -0.14189754E+02 0.22271934E+01 0.64840489E-02 -0.50900690E-06 3
-0.66263206E-09 0.14445464E-12 0.07774954E+05 -0.14189754E+02 4
CH2(S,R) 050595C 1H 2 0 0G 300.00 3000.00 1000.00 1
0.17394471E+01 0.51764320E-02 -0.42153641E-06 -0.53463645E-09 0.11763388E-12 2
0.02478779E+06 -0.11177380E+02 0.17394471E+01 0.51764320E-02 -0.42153641E-06 3
-0.53463645E-09 0.11763388E-12 0.02478779E+06 -0.11177380E+02 4
CH2*(S) 071291C 1H 2 0 0G 300.00 3000.00 1000.00 1
0.17394471E+01 0.51764320E-02 -0.42153641E-06 -0.53463645E-09 0.11763388E-12 2
-0.59624932E+04 -0.12329198E+02 -0.25071146E+01 0.12199585E-01 0.59056043E-06 3
-0.66420434E-08 0.24363521E-11 -0.45017656E+04 0.10931940E+02 4
D 051795C 1 S 300.00 3000.00 1000.00 1
0.16900997E+01 0.11069085E-02 -0.12616481E-06 -0.11996654E-09 0.28811839E-13 2
-0.02996751E+04 -0.09755593E+02 0.16900997E+01 0.11069185E-02 -0.12616481E-06 3
-0.11996654E-09 0.28811839E-13 -0.02996751E+04 -0.09755593E+02 4
G 071291C 1 0 0 0G 300.00 3000.00 1000.00 1
0.16900997E+01 0.11069085E-02 -0.12616481E-06 -0.11996654E-09 0.28811839E-13 2
-0.79270331E+03 -0.10340458E+02 -0.12628431E+01 0.73135700E-02 -0.72650602E-06 3
-0.57274971E-08 0.29829661E-11 0.68074341E+02 0.52818985E+01 4
END
!
! GAS-SURFACE REACTION MECHANISM.
! Incorporating: activation of surface by H atoms; recombination of H on the surface;
! chemisorption of CH3 radicals onto activated sites; incorporation of these molecules into
! the diamond structure (See Reference 8.13).
!
REACTIONS
! New Chlorine Chemistry
CH(S) +CL <=> C(S,R) + HCL 0.5032 0.0 1000.0
STICK
CH2(S) +CL <=> CH(S,R) + HCL 0.5032 0.0 1000.0
STIC
CH3(S) +CL <=> CH2(S,R) + HCL 1.00 0.0 1000.0
STICK
! Initiation Reaction
CH(S) +H <=> C(S,R) + H2 2.14 0.0 7300.0
STICK
! Surface H Replenishing
C(S,R) + H <=> CH(S) 0.3 0.0 0.0
STICK
! Methyl Addition Reactions
C(S,R) + CH3 <=> D + CH3(S) 0.33 0.0 0.0
STICK
CH2(S) + H <=> CH(S,R) + H2 2.14 0.0 7300.0
STICK
CH3(S) + H <=> CH2(S,R) + H2 4.28 0.0 7300.0
STICK
CH2(S,R) + CH(S,R) <=> CH2(S) + CH(S) 6.0E+19 0.0 0.0
! Acetylene Addition Reactions
C(S,R) + C2H2 <=> D + HCCH(S,R) 8.0E10 0.0 7700.0
CH(S) + HCCH(S,R) <=> C(S,R) + H2CCH(S) 6.0E19 0.0 0.0
H2CCH(S) + C(S,R) <=> D + CH2(S)+CH(S,R) 6.0E19 0.0 0.0
CH2(S) + CH(S,R) <=> CH2*(S)+ CH(S,R) 6.0E19 0.0 2122.0
CH(S,R) + H + CH2*(S) <=> CH(S) + CH(S) + H2 2.14 0.0 7300.0
! Carbon-Atom Addition Reactions
C(S,R) + C <=> D + C(S,R3) 0.33 0.0 0.0
C(S,R3) + CH2(S) <=> CH(S,R) + CH(S) 6.0E19 0.0 0.0
CH(S,R) + H <=> CH2(S) 0.3 0.0 0.0
STICK
! Other Radical-Termination Reactions
C(S,R3) + H2 <=> CH2(S,R) 0.3 0.0 0.0
STICK
C(S,R3) + H <=> CH(S,R2) 0.3 0.0 0.0
STICK
CH(S,R2) + H <=> CH2(S,R) 0.3 0.0 0.0
STICK
CH2(S,R) + H <=> CH3(S) 0.3 0.0 0.0
STICK
CH(S,R2) + H2 <=> CH3(S) 0.3 0.0 0.0
STICK
! Graphite Reactions
CH3 + C(R,G) <=> G + CH3(S) 3.0E-2 0.0 0.0
STICK
C + C(R,G) <=> G + C(S,R3) 3.0E-2 0.0 0.0
STICK
C2H2 + C(R,G) <=> G + HCCH(S,R) 3.0E-2 0.0 0.0
STICK
C(R,G) + H <=> CH(S,G) 3.0E-2 0.0 0.0
STICK
CH(S,G) + H <=> C(R,G) + H2 2.14E-2 0.0 7300.0
STICK
CH(S,G) + G + H <=> CH(S,R) + CH(S) 2.14E-1 0.0 7300.0
STICK
END
Figure 8.5. An example of the SURFACE CHEMKIN input file to the Surface Linking File (the surface reactions are taken from Reference 8.13). The symbols R, S, D and G means radical, surface species, diamond and graphite respectively. The numbers which follow the symbol R are the number of unpaired electrons on the surface radical species.
8.4
Application codes
Having executed all the various computer packages and created the relevant linking files, the subroutines are now ready to be called upon by the application codes to simulate the CVD growth process. In the present study two such application codes were used namely the SENKIN and the SPIN application codes.
(a) SENKIN
SENKIN8.14
is a Fortran computer program that computes the time evolution of a homogeneous
reacting gas mixture in a closed system.
The program considers five different problem types:
(a) an adiabatic
system with constant pressure,
(b) an adiabatic system with constant volume,
(c) an adiabatic system with the volume is a specified function of time,
(d) a system where
the pressure and temperature are constant, and
(e) a system where
the pressure is constant and the temperature is a specified function of
time.
In
the case of diamond CVD process, the system employed is that where the pressure
and temperature are held constant, i.e.
case (d). The role of SENKIN in the
modelling of experimental results obtained by MBMS will be discussed in detail
in Section 8.6.
To solve a problem using SENKIN the user must access the CHEMKIN thermodynamic data base, execute the CHEMKIN Interpreter, and link the SENKIN with the CHEMKIN subroutine libraries, pass input data to SENKIN, and store the output data that is produced. Figure 8.6 shows the relationship between these various components.
Figure 8.6. Relationship of the SENKIN program to the CHEMKIN pre-processor
and the associated input and output files (Adapted from Reference 8.13)
The first step is to execute the CHEMKIN interpreter, which reads the user supplied information about the species and chemical reactions for a particular reaction mechanism. It then extracts information from the Thermodynamic Data Base about the species thermodynamic properties. A CHEMKIN link file is then written upon execution of the interpreter which in turn is accessed by the SENKIN program.
The
input that defines a particular problem and the parameters required to solve it
are read by the program SENKIN. The
input data is read using a Keyword format that is entered in the senk.inp file (See Figure 8.7 below).
! SENK.INP
!
! ----------------------------------------Problem Selection
!
CONT
!
! ----------------------------------------Initial Conditions
!
PRES 0.026
TEMP 2000
REAC H2 0.99
REAC CH4 0.01
!
! ----------------------------------------Integration Control
!
TIME 5.0
DELT 2.0E-02
ATOL 1.0E-09
RTOL 1E-05
!
END
Figure 8.7. An example of the input data that is read using a Keyword format
by SENKIN.
The keyword CONT will
produce a solution with the temperature and pressure held constant at the
initial values, and the equations solved will be those of case (d). In the above example (Figure 8.7), the
initial conditions are those of an isothermal gas mixture of 1% methane (REAC
CH4) in hydrogen (REAC H2) at 20 Torr - the units used in
the input file is in atmospheres (PRES 0.026) and Kelvin (TEMP 2000). This gas mixture is allowed to react for 5
seconds (TIME 5.0), meaning that the time integration will proceed from time
zero until this final time. A more
detailed description of the keywords used in SENKIN can be found in Reference
8.14.
The output from program SENKIN is written in two forms: text and binary. Text output is written to display the outcome of the execution on a terminal, while the binary file restores a copy of the run. The information written to both text outputs include a summary of the initial conditions and printouts of the partial solution at a selected time interval. The full solution is written in the binary file which is useful in providing initial data for restart calculations. From this output the species concentrations may be obtained, by executing a program called DATAC.EXE which converts mass fractions written in the SENKIN output file into species mole fractions. The final result is therefore species mole fractions as a function of reaction time. An example of this is shown in Section 8.5.
(b) SPIN
SPIN8.15 is one of the application codes
supplied with the CHEMKIN software package which construct and solve a system
of differential equations describing a physical problem (in the case of SPIN, a
CVD reactor), using standard subroutines from various modules which together
form the complete simulation package.
Thus SPIN simulates the flow of chemically reacting fluid towards a
growth surface on which gas-phase species can react. The gas-phase chemistry is modelled by a set of chemical
reactions with rate constants provided by the gas-phase CHEMKIN module. The transport properties of the gas-phase
species are continually evaluated by the TRANSPORT fitting module. Finally the production/consumption of
gas-phase species on the growth surface is accounted for by a set of surface
chemical reactions with rate constants calculated by the SURFACE CHEMKIN module
(See Figure 8.4). Figure 8.8 shows an
example of a typical SPIN input file.
! SPIN.INP Figure 8.8. Input file of the SPIN application code.
! Input file for SPIN application code. Full details of the different keywords used is found
! in Reference 8.15.
!---------------Problem type
!
COMP
!NOFT
STAG
!TGIV
ENRG
!RSTR
!
!---------------Solution Method Options
!
!USTG
!
!---------------Newton iteration controls.
!
ATOL 1.E-8
RTOL 1.E-3
NJAC 20
!
!---------------Time step controls.
!
ATIM 1.E-8
RTIM 1.E-3
TIME 100 1.E-6
TIM2 100 1.E-6
UFAC 2.0
DFAC 2.2
DTMN 1.E-10
DTMX 1.E-4
TJAC 20
ISTP 100
IRET 10
!
!---------------Solution bound controls.
!
SFLR -1.0E-04
SPOS 1.E-13
!
!---------------Grid control parameters.
!
NPTS 20
NMAX 65
!JJRG 90
!PCAD 0.75
!RGTC 1.0
GRAD 0.9
CURV 0.9
GRID 0.00
GRID 0.02
GRID 0.04
GRID 0.06
GRID 0.08
GRID 0.10
GRID 0.12
GRID 0.14
GRID 0.16
GRID 0.18
GRID 0.20
GRID 0.22
GRID 0.24
GRID 0.26
GRID 0.28
GRID 0.30
GRID 0.32
GRID 0.34
GRID 0.36
GRID 0.38
GRID 0.40
!
!---------------Initial guess & reactor operating
! conditions.
!
PRES 0.026
TINF 2173
TDSK 1173
!RADB
AINL 0.0
OINL 0.0
UINF 1.0
XSTR 0.0
XCEN 0.2
XEND 0.4
WMIX 0.2
TEMP 0.0 2173
TEMP 0.4 1173
!
!----------------Set gas concentration at in-let
!
REAC H2 0.9429
REAC H 0.0471
REAC CH4 0.01
!
!----------------Set initial guess of gas concentrations at disk
!
PROD H2 0.990041
PROD H 0.723930E-06
PROD CH4 0.991787E-02
PROD CH3 0.162266E-06
PROD CH2 0.902875E-13
PROD CH 0.317941E-18
PROD C 0.135916E-20
PROD C2H6 0.656252E-07
PROD C2H5 0.246341E-10
PROD C2H4 0.119802E-04
PROD C2H3 0.522252E-10
PROD C2H2 0.285896E-04
PROD C2H 0.109323E-14
!
!----------------Set initial guess of surface concentrations
!
!SURF CH(S) 9.51E-01
!SURF C(S,R) 4.87E-02
!SURF HCCH(S,R) 6.11E-15
!SURF CH2(S) 3.98E-04
!SURF CH(S,R) 2.23E-05
!SURF C(R,G) 1.00E-04
!SURF CH(S,G) 1.00E-04
!SURF H2CCH(S) 1.16E-14
!SURF CH3(S) 1.63E-06
!SURF CH2(S,R) 1.00E-07
!SURF CH2*(S) 1.64E-04
!SURF C(S,R3) 1.05E-08
!SURF CH(S,R2) 1.00E-07
!
!----------------Set activity of bulk phases
!
!ACT D 0.9999
!ACT G 1.0E-04
!
!---------------Multicomponent transport options.
!
MULT
!TDIF
!TRCE
!REOR
!GFAC 1E-04
!SFAC 1E-04
!
!---------------Chemistry control options.
!
!---------------Printing options.
!
PRNT 2
!
!---------------Miscellaneous.
!
END
~
A full description of the keywords used in the SPIN input file is shown in Reference 8.15. From the SPIN output the species concentrations may be obtained, by executing a program called SPDAT.EXE which converts mass fractions written in SPIN output file into species mole fractions. The final result is species mole fractions as a function of distance from (in this case) the filament to the substrate surface. An example of this is shown in Section 8.5.
8.5
Numerical simulation of the diamond CVD process
The first time that we attempted to run the SPIN simulation code, it crashed reporting that the matrix, which is constructed to solve the differential equations governing the mathematical problem, was singular. A considerable amount of time was spent breaking the problem into a number of simpler problems which could be analysed in greater detail in order to find the origin of the singularity.
In
order to test the formulation of the gas-phase part of the problem in
isolation, a simpler problem was examined involving only the gas-phase
reactions, using the SENKIN application code.
This code considers an isothermal gas mixture maintained at constant
pressure and temperature (Section 8.4 (a) ) and calculates how the composition
of the mixture changes as a function of time as a result of the chemical
reactions. The results that were
obtained for a mixture of 0.3% CH4 in H2 held at 2000K
(Figure 8.9) were very similar to results reported by Harris et al.8.2 This confirmed that the gas-phase CHEMKIN
module was working correctly.
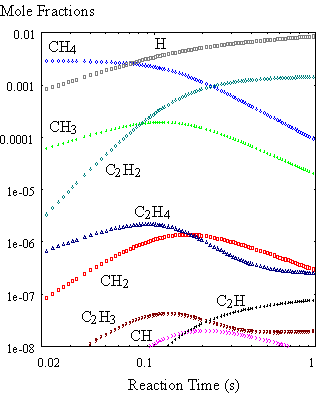
Figure 8.9. Calculated time history of gas
composition. The calculation is carried
out using the SENKIN code for isothermal conditions at 2000K for an initial
source gas mixture of 0.3% CH4 in H2 (99.7%).
It
was later discovered that the SPIN application code could be run with empty
surface reaction files (non existent surface reaction files cause it to crash),
without it running into singularity problems.
In this way, the flow of a chemically reacting fluid towards a
chemically inert surface could be simulated, making it possible to check that
the fluid flow was being modelled correctly, using the transport properties
provided by the TRANSPORT fitting module and based on the description of the
gas-phase chemistry provided by the gas-phase CHEMKIN module, but ignoring the
surface chemistry. Such simulations may
be compared to the results obtained by the MBMS system, in which there is
currently no growth surface in the vicinity of the sampling cone. The results obtained for this simulation
(Figure 8.10) showed similar trends to those reported by Dandy et al.8.5
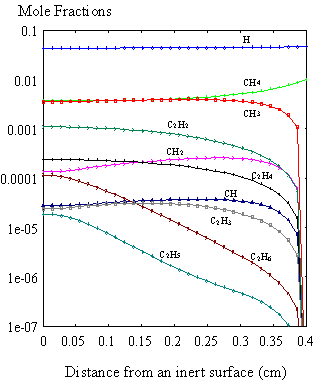
Figure 8.10. Mole fraction profiles calculated for 1% CH4
in H2 at 20 Torr using the SPIN application code, in the absence of
surface reactions. Initial deposition
conditions are: filament temperature 2000K, filament/substrate distance
4 mm (or 0.4 cm). In the
above illustration, the surface is at 0 cm, and the filament is at
0.4 cm.
The
next step was performed solely by Jonathan Cole8.16, who used the
SURFTHERM programming module to examine in detail the thermodynamic and kinetic
data which must be provided for all surface species and surface reactions. Some of this data must be input in a special
format and SURFTHERM can be used to produce tables of values over various
temperature ranges: errors are revealed by the appearance of physically
unreasonable values. In order to
confirm that the SURFACE CHEMKIN module was working correctly, Cole used
another application code called SKSAMPLE.
SKSAMPLE models an idealised system consisting of a completely uniform
and isothermal gas mixture lying above a chemically reactive surface. As there are no temperature or concentration
gradients in the system, there is no fluid flow and the transport properties
may be ignored. The net production and
consumption rates of the various gas-phase and surface species are calculated
from the kinetic data supplied by the gas-phase CHEMKIN and SURFACE CHEMKIN modules
and SKSAMPLE predicts how the gas-phase composition and surface site fraction
change with time, starting from a specified gas-phase condition. Again, SKSAMPLE could be executed
successfully and gave physically plausible results.
Having ascertained that the various parts of the complete problem were correctly formulated, it appears that the mathematical singularities may have arisen because the initial guess was very far from the final solution, making the system ‘ill-conditioned’. Eventually, a search strategy was identified for conditioning the initial guess in such a way as to bring it within the domain of convergence of the computation. The initial guess may be conditioned by forcing the SPIN code to perform a ‘time evolution’, whereby the original system of equations is augmented by a time-dependent term to yield a time-dependent system, which mathematically is much less susceptible to divergence problems. The time-dependent solution is then integrated to give a much better guess to the solution of the steady-state problem. Computationally, this is more expensive and time-consuming than solving the steady-state problem from the start, but it is more reliable. A similar strategy is followed automatically by SPIN when less severe divergence problems are encountered, but the singularities found in the present simulation prevented it from even ‘getting off the ground’.
In this way the full problem,
including surface reactions, was solved and the results of the simulation of a
hot filament CVD reactor are shown in Figure 8.11. These also show similar trends to those reported by Dandy et al.8.5
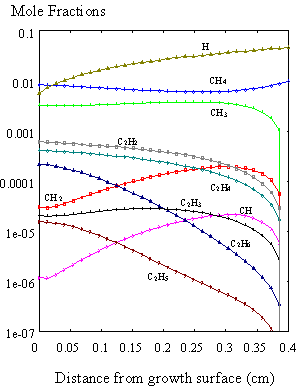
Figure 8.11. A full simulation of the diamond CVD growth
process using SPIN. The species mole
fractions are presented with surface reactions included in the calculations.
8.6 Numerical simulation of the gas-phase
composition vs. filament temperature
for 1% CH4 in H2 at 20 Torr
As
previously mentioned in Section 8.5, It is possible to run the SPIN application
code with empty surface reaction files, because such simulations may be
compared to the results obtained by the MBMS system. This application code was therefore used in order to test the
validity of the MBMS data. Unlike the
work performed by the various groups using the CHEMKIN package,8.2-8.7,8.9,8.13
for the first time a numerical simulation of the gas-phase composition using a
1% CH4 in H2 gas mixture as a function of filament
temperature was performed, in precisely the same way in which the MBMS result
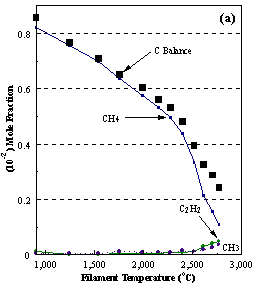
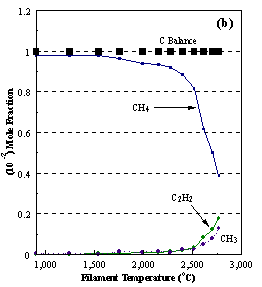
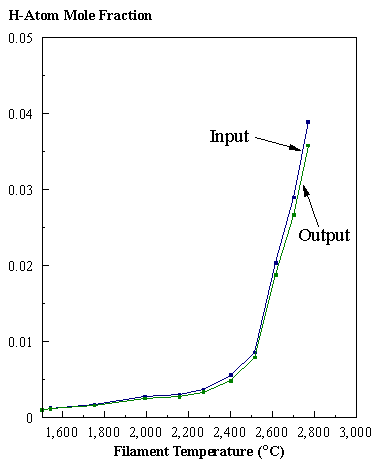
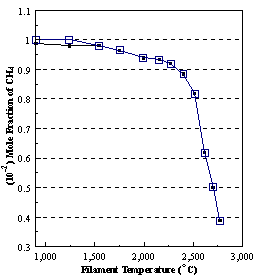
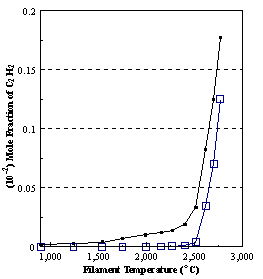
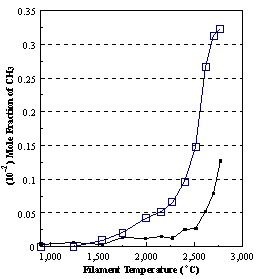
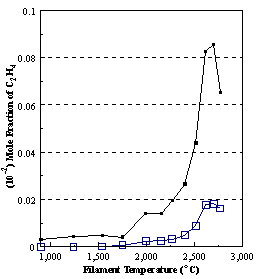
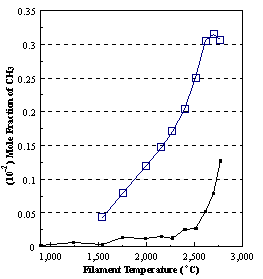
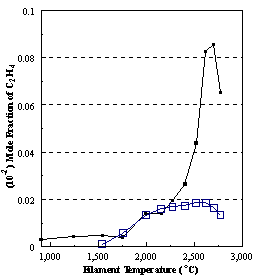
are presented. Figure 8.12 shows how
the distribution of the major observable stable gas-phase species [CH4
(m/e = 16), C2H2
(m/e = 26)] and methyl radicals (m/e = 15), measured by MBMS, vary as a
function of filament temperature for an initial feedstock of 1% CH4
in H2 measured 4 mm from the filament. The data is presented in two forms:
(a) with no thermal diffusion corrections being made, and (b) with thermal
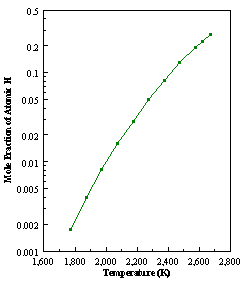
diffusion corrections being made in the calculations. Note that the carbon balance, defined as (total C fraction
measured)/(C fraction in the feed gas) and shown as a black filled square in
Figure 8.12 (b), is constant (and equal to one) throughout. In the present work, the SPIN application
code was performed without including the thermal diffusion keyword in the input
file, and so we are simulating the variation in the concentrations of the
hydrocarbon species as a function of filament temperature shown Figure 8.12
(b).