
|
|
Laser Raman Spectra have been obtained for crystalline adamantane, diamantane, cyclohexamantane and diamond, using parallel and cross polarisation filters. The spectra are presented in JCAMP format to allow on-line interaction with the data by the reader. The various peaks in the spectra are compared to those often seen from chemical vapour deposited (CVD) diamond films, and which have been assigned to 'nanophase' diamond. Although there is a superficial correlation between the peak positions for the polymantanes and those seen in CVD diamond, the conclusion is that the nanophase diamond peaks are not an intrinsic vibrational mode of a polymanatane, but instead must be a result of some other aspect of the nanophase scale of the films, such as surface modes.
Laser Raman Spectroscopy (LRS) is an important technique used to analyse solid state materials [1]. Recently it has been extensively utilised to determine the quality of diamond films deposited by chemical vapour deposition (CVD) [2-6,12], since the ratio of the characteristic sharp diamond peak at 1332 cm-1 to that of the broad feature at ~1550 cm-1 corresponding to the graphitic G-band is an indication of the phase purity of the sample [2]. In some LRS studies of poorer quality diamond or of the initial stages of diamond growth, several peaks around 1130-1230 cm-1 have been observed [7], and have been tentatively assigned to 'nanophase' diamond, a term used to describe a material consisting of carbon having short range crystal order surrounded by a matrix of less ordered material. However these peak assignments are still controversial and more data are needed to clarify the situation. Apart from the diamond peak, the major peaks seen in this wavelength range are 1130 cm-1 [17], 1140 cm-1 [18] and 1149 cm-1 [7], all assigned to nanophase diamond, 1467 cm-1 [7] and 1480 cm-1 [17] assigned to a 'diamond precursor' or polyacetylene, and 1200-1210 cm-1, which has been seen in highly defective films grown by hot filament CVD [19], in highly B-doped diamond films [20], and at the substrate interface after hetereoepitaxial growth of diamond on Si [21], again being assigned to nanophase diamond. Confocal Raman studies of heteroepitaxial diamond film growth on Pt substrates [22] revealed peaks at 1230, 1640 and 1470-90 cm-1 assigned to defective disordered diamond nanocrystals, 1400 and 1530 cm-1 assigned to disordered graphite, and 1500 cm-1 assigned to a distorted sp3-bonded carbon network. Very recently, Prawer et al [8] obtained the LRS spectrum of so called 'amorphous diamond' (an randomly arranged but fully sp3-bonded carbon network) and found that it was dominated by a large peak at around 1200 cm-1.
The nanophase diamond that allegedly gives rise to some of these peaks can be considered to be a collection of fused adamantane units, since adamantane, C10H14, is the smallest building block exhibiting the diamond structure (see Fig.1). Higher diamondoids in the series exist [9], continuing with diamantane, C14H20, triamantane, C18H24, and so on. The first three in this series have been studied using LRS and the resulting peaks assigned to either C-C or C-H vibrational modes [10,13]. However these reports were performed before diamond CVD was established, and so spectra are not available in the literature which cover the wavenumber region of interest to diamond CVD researchers. This is partly the motivation for the present work - to use modern LRS systems to obtain the spectra of relevant diamondoid structures over an appropriate wavelength range, and to compare these results with features observed in diamond films. We have chosen to use the first two in the series, adamantane and diamantane, since these are readily commercially available, as well as cyclohexamantane, C26H30, which is a diamondoid that has only recently been extracted and isolated from petroleum [11].
|
|
|
Adamantane and diamantane (also known as congressane) were purchased in crystalline form from BDH and Aldrich Chemicals, respectively. The cyclohexamantane samples were two small crystals (0.5 mm on a side) obtained from J. Dahl of Stanford University by extraction from petroleum oil [11]. The single crystal diamond sample was a type Ib high pressure high temperature (HPHT) synthetic diamond from Sumitomo Industries. This crystal measured 4x4x2 mm and contained around 50-100 ppm of singly-substituted nitrogen.
Laser Raman spectroscopy was performed using a Renishaw Raman System 1000 spectrometer operating at excitation wavelengths of 488 and 514.5 nm in a backscattered configuration. The laser was linearly polarised, and a polarisation filter and quarter wave plate were placed in the analyser beam path to give information about the symmetry of the vibrational modes in the samples.
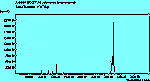
The various spectra are shown in Table 1. The JCAMP file (.jdx) is an interactive file which can be viewed using a suitable reader, such as the Chime plug-in. This will allow the reader to zoom in and out of the spectra, and to save the data as x,y coordinates to their own computer. If the appropriate software to view these files is not available, the important parts of the spectra can still be see as GIF images, which can also be printed if required.
| Sample |
JCAMP File |
Full Range |
Gif Images 400-1500 cm-1 |
2600-3000 cm-1 |
|---|---|---|---|---|
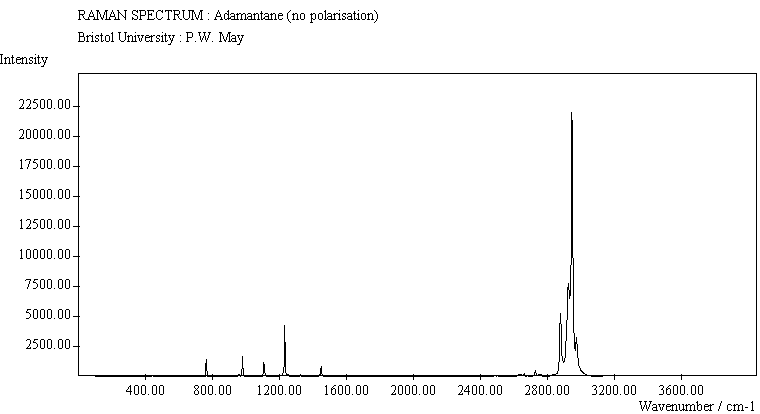
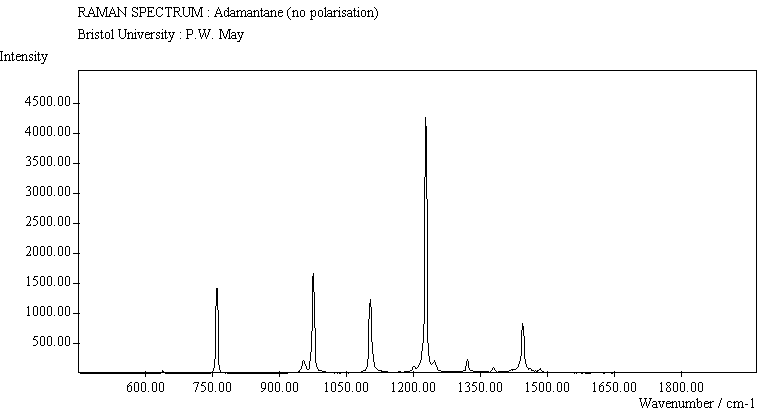
| Adamantane (no pol.) |
 |
 |
 |
 |
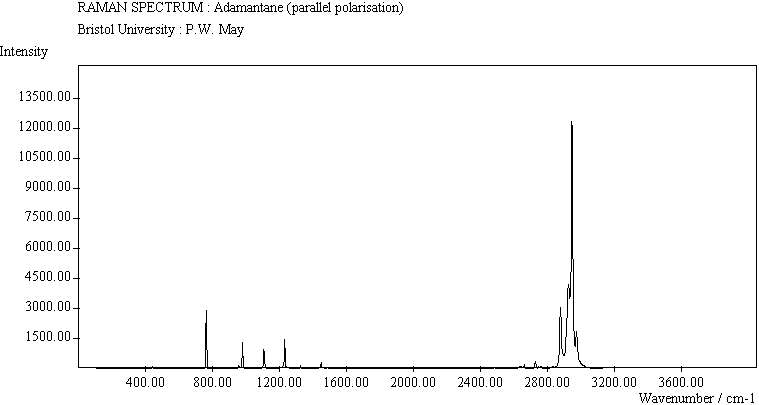
| Adamantane (para. pol.) |
 |
 |
 |
 |
| Adamantane (cross pol.) |
 |
 |
 |
 |
| Diamantane (no pol.) |
 |
 |
 |
 |
| Diamantane (para. pol.) |
 |
 |
 |
 |
| Diamantane (cross pol.) |
 |
 |
 |
 |
| Cyclohexamantane (no pol.) |
 |
 |
 |
 |
| HPHT Diamond (no pol.) |
 |
 |
||
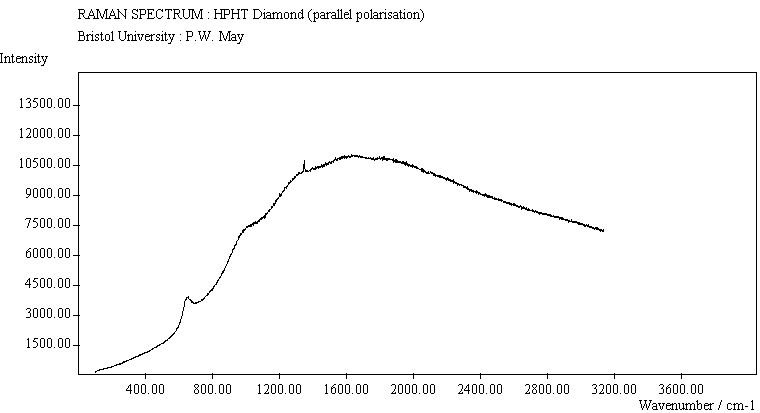
| HPHT Diamond (para. pol.) |
 |
 |
||
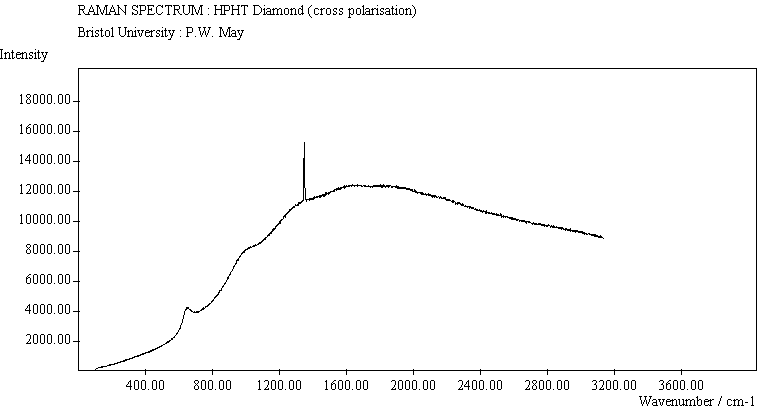
| HPHT Diamond (cross pol.) |
 |
 |
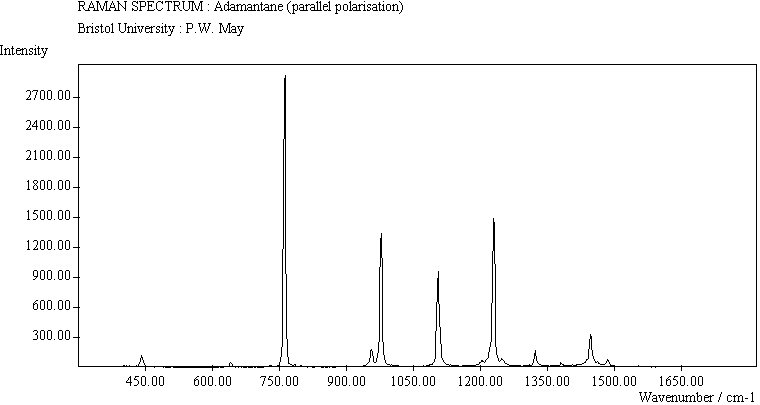
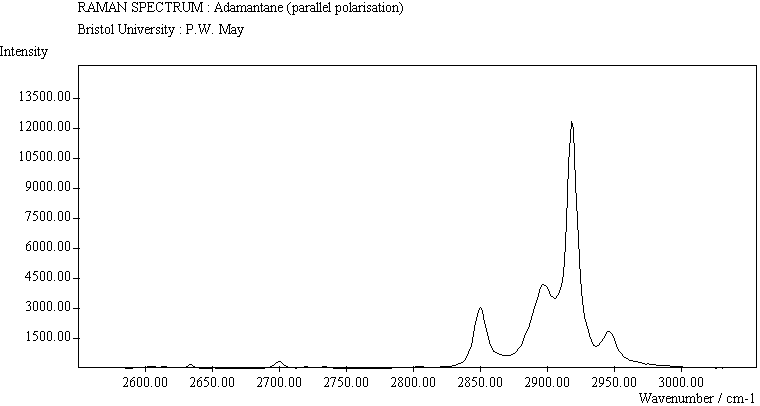
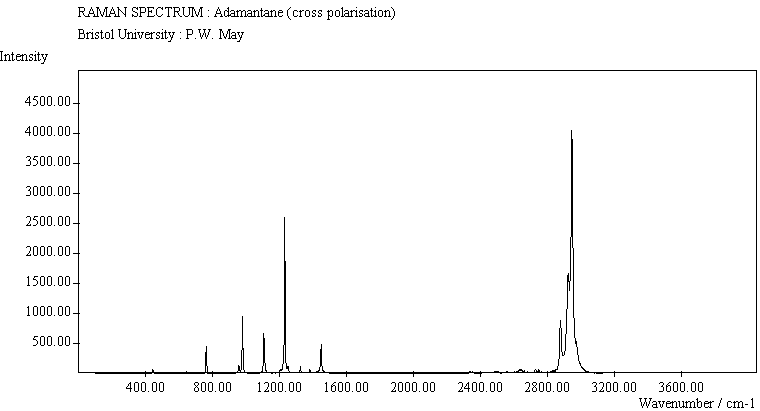
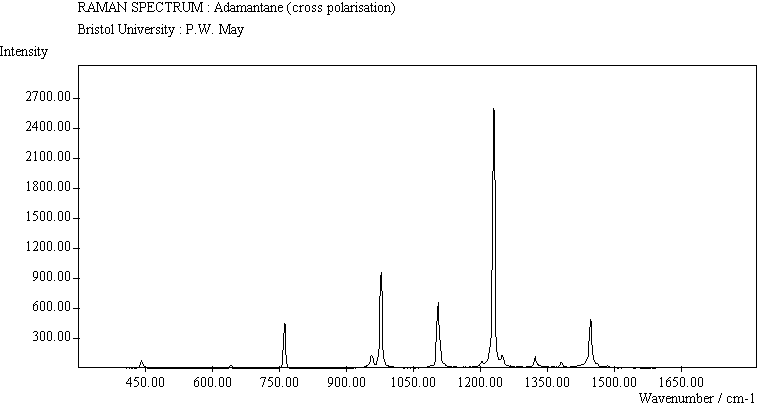
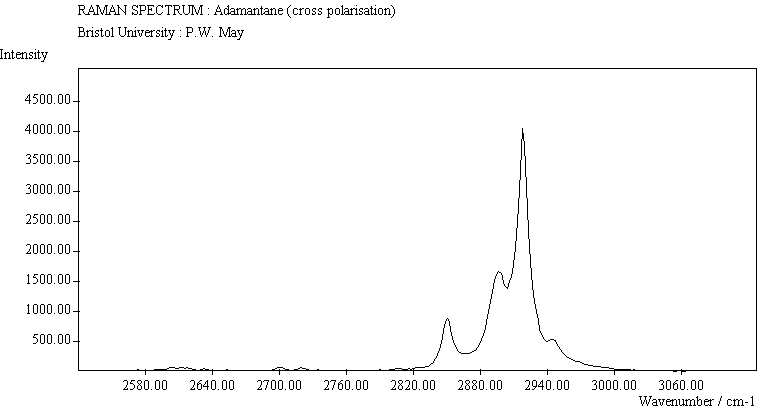
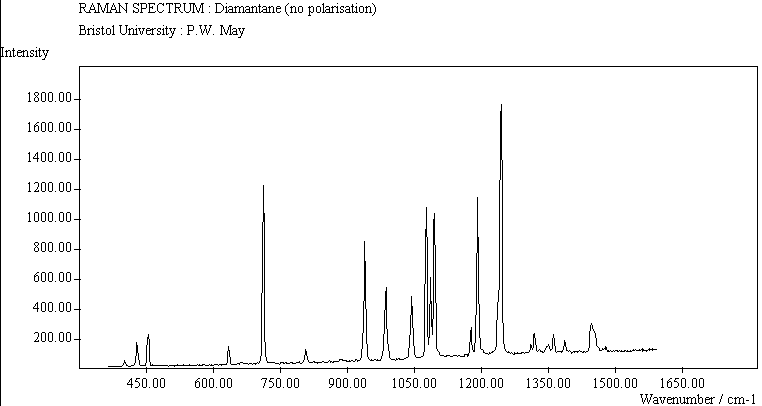
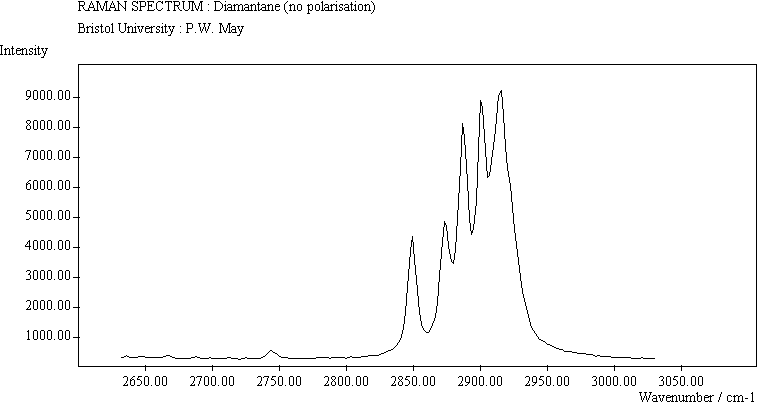
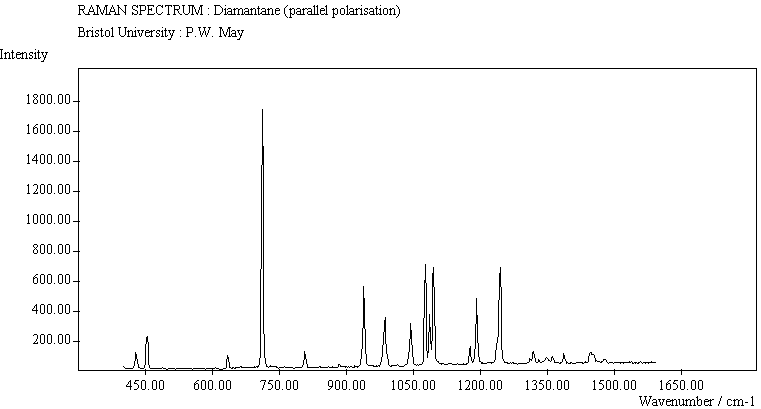
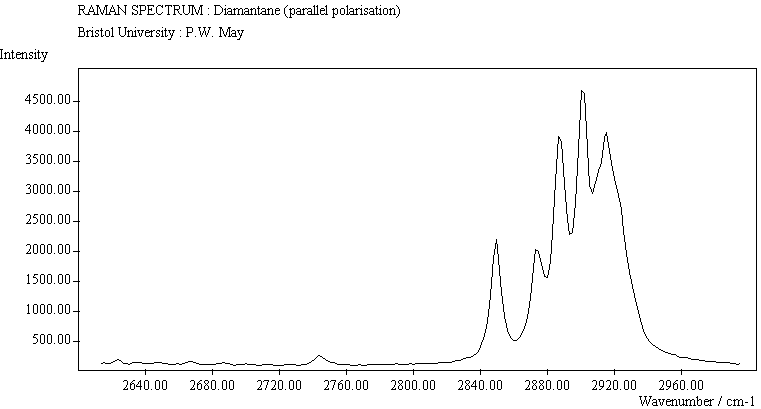
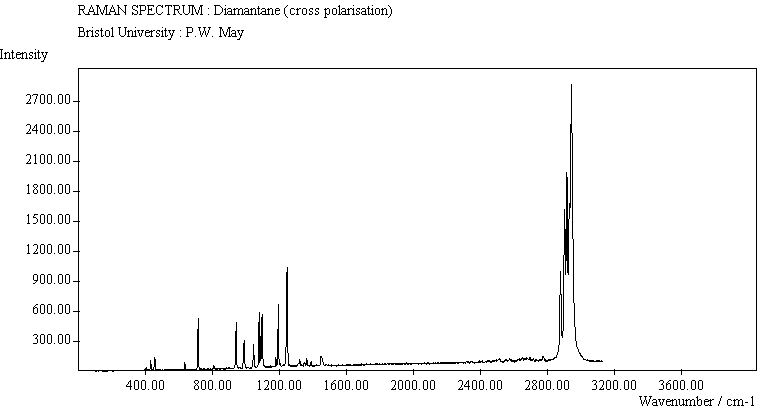
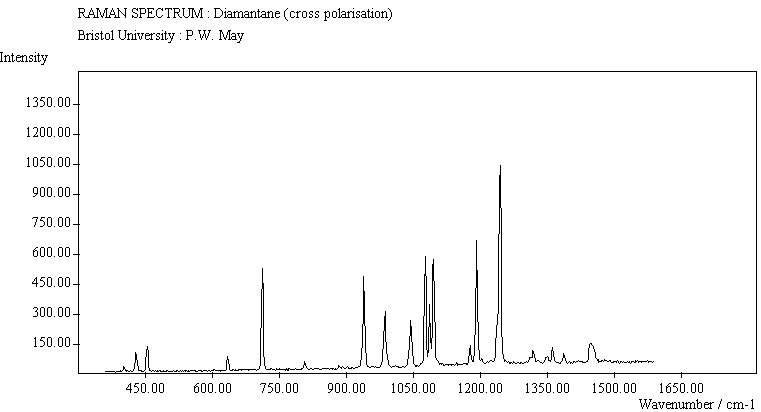
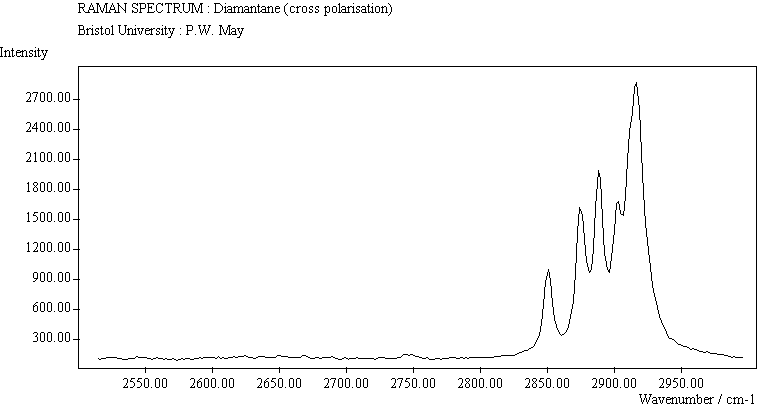
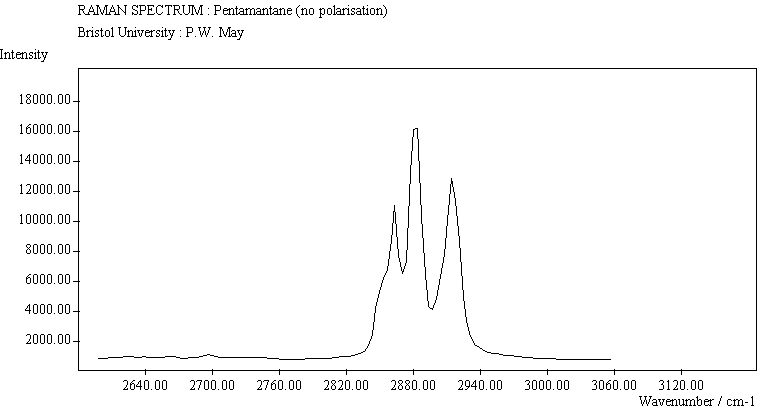
The spectra for the polymantanes show two regions, one at low energy (roughly 300-1600 cm-1) corresponding to the various C-C skeletal bends, rocks and stretching modes, and one at high energy (~2600-3000 cm-1) corresponding to the C-H stretching modes. Using the 3N-6 rule for the number of vibrational degrees of freedom: adamantane has 24 atoms = 66 modes, diamantane has 34 atoms = 96 modes, and cyclohexamantane has 56 atoms = 162 modes, although, of course, only some of these will be Raman active. The various modes for adamantane and diamantane are given in ref.[10]. The modes of cyclohexamantane are classified as 18(A1g+A1u) + 9(A2g+A2u) + 27(Eg+Eu), of which only the A1g and Eg modes are Raman active.
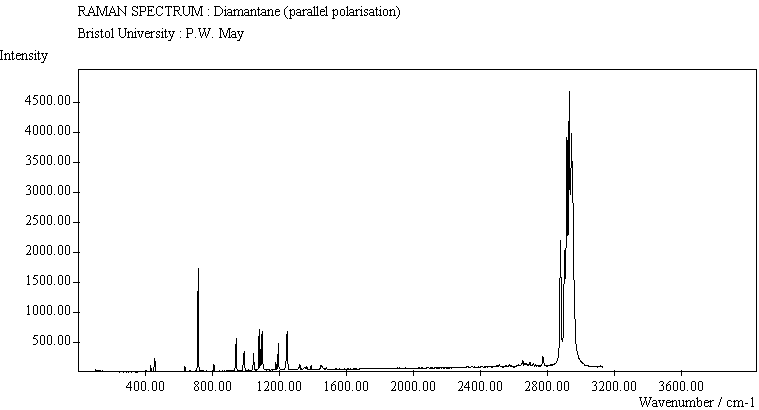
The peak assignments for adamantane and diamantane have been taken from ref.[10] and are reproduced in Table 2. Our spectra agree with these very well, with the peaks appearing to within 1 cm-1 of those seen in this reference. There are some differences, though, especially at lower energies. Our spectra reveal some lines which were only present at low temperature in ref.[10]. This could be because we used a higher energy laser for the excitation, or that our detector was more sensitive at these lower energies. We have also seen a number of peaks between 1472-184 cm-1 which haven't been reported before and so have yet to be assigned. These peaks could be a result of second order effects. By comparing the relative heights of the various peaks to their neighbours for different polarisations, we can see that for adamantane, the peaks at 760 cm-1 and 1218 cm-1 are strongly polarised, as are all the C-H peaks between 2800-3000 cm-1. For diamantane we see that the peaks at 709, 1181 and 1235 cm-1 are strongly polarised, as well as the C-H peaks between 2800-3000 cm-1.
| Mode | Adamantane | Diamantane | Cyclohexamantane |
|---|---|---|---|
| CCC bend | 340 | ||
| 349 | |||
| 398 | 401 | 396 | |
| 404 | 428 | ||
| 441 | 453 | 445 | |
| 498 | |||
| 637 | 631 | 654 | |
| 674 | |||
| 680 | |||
| CC stretch | 708 | ||
| 758 | |||
| 802 | 817 | ||
| CH2 rock? | 864 | ||
| CC stretch | 950 | 933 | |
| 923 | |||
| CH2 rock | 971 | ||
| CCC bend | 979 | 986 | |
| 1037 | 1034 | ||
| 1050 | |||
| 1068 | 1072 | ||
| 1078 | 1076 | ||
| HCC bend | 1097 | 1086 | |
| 1167 | 1141 | ||
| 1193 | 1182 | 1183 | |
| 1206 | |||
| 1220 | 1230 | 1221 | |
| 1238 | 1234 | 1258 | |
| 1312 | 1299 | 1293 | |
| 1307 | 1311 | ||
| 1338 | 1336 (or the 'diamond' peak??) | ||
| 1349 | 1352 | ||
| 1369 | 1375 | 1376 | |
| HCH bend | 1434 | 1433 | 1444 |
| 1465 | |||
| CCC scissor | 1449 | ||
| 1472 | |||
| unknown | 2268 | ||
| 2312 | 2297 | ||
| 2326 | |||
| 2388 | |||
| 2414 | |||
| 2454 | |||
| 2476 | 2483 | 2469 | |
| 2531 | 2509 | ||
| 2547 | 2545 | ||
| 2571 | |||
| 2604 | 2597 | ||
| 2613 | 2623 | 2624 | |
| 2633 | 2635 | ||
| 2651 | 2647 | 2641 | |
| 2667 | 2663 | ||
| 2687 | |||
| 2700 | 2697 | 2695 | |
| 2717 | 2712 | 2716 | |
| 2731 | 2724 | 2737 | |
| 2755 | 2743 | ||
| 2804 | |||
| CH stretch | 2847 | 2846 | 2848 |
| 2871 | 2860 | ||
| 2884 | 2878 | ||
| 2893 | 2898 | 2903 | |
| 2915 | 2911 | 2911 | |
| 2941 |
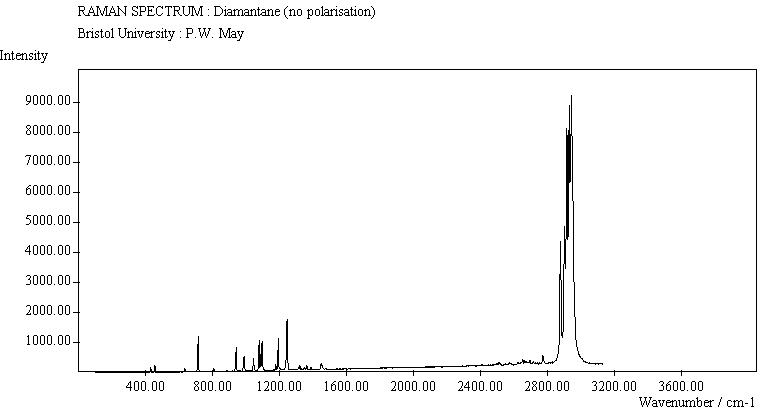
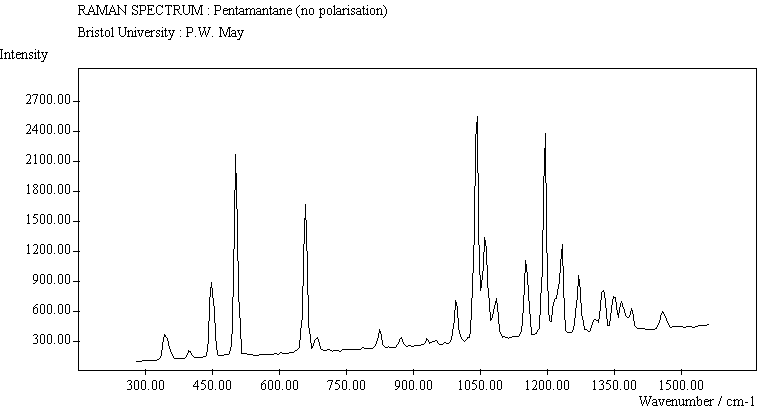
For cyclohexamantane, tentative assignment of peaks has been suggested in ref[11] and are also reproduced in Table 2. As expected, we see many more peaks, but unfortunately no polarisation experiments were performed on these samples XXXXX.
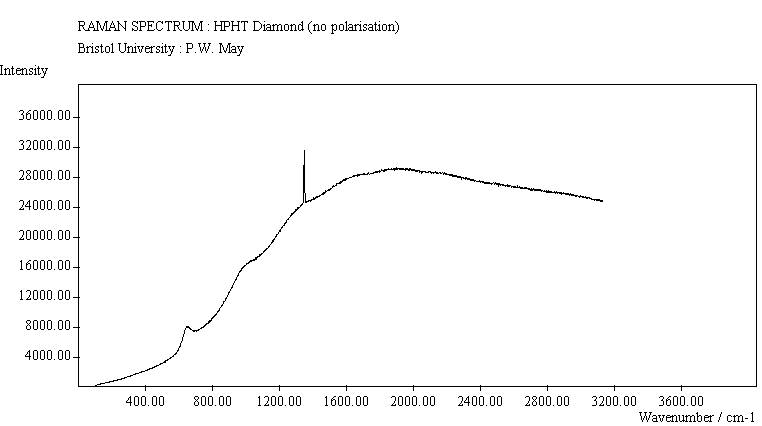
The diamond spectra show the large photoluminescence (PL) background often seen in Raman spectroscopy of diamond. Only one sharp, strongly polarised line is seen, at 1333 cm-1, characteristic of the diamond fully symmetric stretching mode. A smaller broad feature at 630-650 cm-1 is a PL feature associated with singly substituted nitrogen.
The polymantane spectra exhibit many peaks in the wavenumber region where nanophase diamond peaks have been reported, and as the size of the diamondoid structure increases the number and complexity of the spectral lines also increases. There is some correlation between the peaks assigned to nanophase diamond and those seen for the diamondoids, but the match is not convincing. For example there is a small cyclohexamantane peak at 1141 cm-1 which is within the range 1130-50 cm-1 for which peaks were seen in nanophase diamond [18] (although no large peaks in the wavelength range are seen for adamantane nor diamantane). Around 1200-50 cm-1, adamantane, diamantane and cyclohexamantane all have peaks (at 1219, 1234 and 1221 cm-1, respectively), and again between 1430-80 cm-1 (1434, 1434 and 1444 cm-1, respectively). But if these are the lines assigned to nanophase diamond, we would expect to see many of the other, stronger, diamondoid lines as well (such as peaks around 1030-90 cm-1), rather than just one or two. This suggests that the spectral lines seen in diamond films are not due to the intrinsic vibrational modes of a diamondoid structure. This leads to the conclusion that some other aspect of the nanophase scale of the films is responsible for the observed features. One possibility is surface phonons and/or edge effects [16], which also have recently been found to have peak positions within the correct wavelength ranges [16].
PWM and TP wish to thank the Royal Society for funding. CDOP and TJD also wish to thank Renishaw UK, DERA malvern, and the EPSRC for financial support.