Ewa Chudyk

I was awarded an MSc in Biotechnology from the Wroclaw University of Technology (Poland) in 2009, where I completed my Masters project in the Molecular Modelling and Quantum Chemistry Group of Professor Sokalski on catalytic activity of fatty acid amide hydrolase. As I was always interested in drug design by both experimental and computational methods, I gained some professional experience in those fields. First I was a Leonardo da Vinci intern in Omnia Molecular Ltd. (Parc Cientific de Barcelona, Spain), studying the problem of antibiotic resistance "on the bench", and then I spent 3 months as a summer student in the BioMolecular Modelling Lab at Cancer Research UK (London Research Institute), developing software to predict protein-protein interactions. I started my PhD studies in October 2009 in the Computational Enzymology Group of Professor Mulholland, where my research area is understanding the catalytic activity and inhibition of fatty acid amide hydrolase and beta-lactamases, as well as investigating reaction modelling methods based on Monte Carlo algorithms.
Catalytic Activity and Inhibition of Fatty Acid Amide Hydrolase (FAAH)
FAAH is a mammalian enzyme mainly responsible for the hydrolytic deactivation of neuromodulators such as anandamide or oleamide. It has previously been found that conformational fluctuations in the FAAH active site affect the reaction barrier for the acylation step with oleamide as a substrate. Geometrical descriptors that give an empirical explanation exist, however a first principles understanding of this phenomenon was missing.
With this in mind, we analysed the changes in the enzyme during reaction with the differential transition state stabilization (DTSS) approach [1], in order to identify active site residues responsible for lowering the activation energy barrier. The pair-wise model was confirmed by calculations in the supermolecule environment, and a variation-perturbation interaction energy decomposition scheme was used to examine the physical nature of catalytic effects, ranging from multipole expansion of the electrostatic terms up to the MP2 level of theory. Wherever electrostatic effects were dominant, these results could be generalised in the form of catalytic fields.
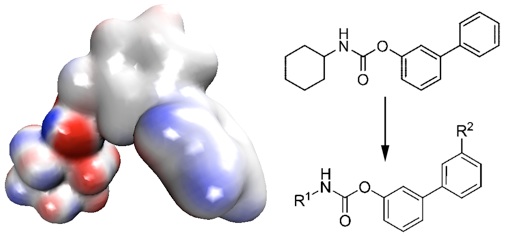
Moreover, we applied analogous DTSS analysis for three covalent, carbamate-based inhibitors: URB524, URB618 and URB694, providing an insight into inhibitor (potential drugs) design. [2]
This project was realised in collaboration with the Molecular Modelling and Quantum Chemistry Group at the Wroclaw University of Technology, Poland (http://ichfit.ch.pwr.wroc.pl/?q=node/10) and the Medicinal Chemistry Group at the University of Parma, Italy (http://www.unipr.it/arpa/dipfarm/medchem/index.htm).
Catalytic field generated around the URB524 inhibitor allows prediction of the most reactive regions of the compound. This was in agreement with a previous structure-activity relationship study described in [2], where positions R1 and R2 were substituted.
Combined theoretical and experimental study on specificity of new β-lactamases
β-lactamases are enzymes produced by Gram-negative pathogens such as Acinetobacter baumannii, Klebsiella pneumoniae or Escherichia coli, and are responsible for cleavage of β-lactam rings in penicillins, cephalosporins and carbapenems. This drug inactivation leads to antibiotic resistance, which has become a real problem in medicine. Of interest in this project is the reaction catalysed by D-class β-lactamases, which proceeds via a covalent aceloenzyme intermediate that separates the acylation and deacylation hydrolysis steps. [3]
Understanding reaction mechanism is a key step in rational drug design. The potential of computational chemistry methods being used in close collaboration with experiments is that these will become useful tools in the investigation of the conformational and dynamical effects investigation for enzymes catalysis. Of special interests for us here are quantum mechanical / molecular mechanical (QM/MM) [4] and molecular dynamics (MD) simulations, as well as crystallography, IR and fluorescence spectroscopy techniques.
Another positive aspect of this theoretical-experimental connection is the development and validation of computational methods to calculate vibrational spectra of enzyme complexes. This might improve structural analysis of spectroscopic outcomes and become a universal tool linking experimental and computational biophysical techniques.
This project is realised in collaboration with experimental group of Dr. Jim Spencer at Dept. of Cellular & Molecular Medicine, University of Bristol (http://www.bristol.ac.uk/cellmolmed/staff/spencer.html).
X-ray crystallographic structure of CTX-M-9 S70G (A-class β-lactamase) in complex with benzylpenicillin (PDB: 3HUO, chain A)
Developing Monte Carlo methods for modelling reaction mechanisms
Combined quantum mechanics/molecular mechanics (QM/MM) simulations provide a useful tool for the investigation of the mechanism of enzyme-catalyzed reactions. Sampling of the configurational space of the system is typically performed using molecular dynamics methods, however due to its limitations further method development is needed, involving e.g. Monte Carlo (MC) algorithms. Encouraged by previous QM/MM MC free energy calculations for the perturbation of water into methane in explicit water [5], we applied the method to an enzyme-catalyzed reaction.
The first model system was a chorismate to prephenate conversion, catalyzed by chorismate mutase. This relatively simple, single-step reaction, proceeding in the same way in both, gas-phase and solution, has been investigated in detail using QM/MM MD simulations [6]. With this as a reference system, we investigated MC sampling along a reaction coordinate for gas-phase, water and enzyme reactions. A novel method was developed that used different z-matrix templates as the reaction proceeded, ensuring better structural sampling during the conversion of the reactant into the product geometry.
In terms of future work, we are planning to extend the method to more complex enzymatic reactions, involving e.g. proton transfer, such as in fatty acid amide hydrolase.
Structures comparison of MD (in blue, the reference system) and MC (in red, generated using 3 z-matrix templates approach) reaction pathway sampling of chorismate into prephenate conversion in gas-phase.
References:
[1] B. Szefczyk, A. J. Mulholland, K. E. Ranaghan, W. A. Sokalski, J. Am. Chem. Soc. 2004, 126: 16148-16159
[2] M. Mor, A. Lodola, S. Rivara, F. Vacondio, A. Duranti, A. Tontini, S. Sanchini, G. Piersanti, J.R. Clapper, A.R. King, G. Tarzia, D. Piomelli, J. Med. Chem. 2008, 51: 3487
[3] E.P. Abraham, E. Chain, Nature 1946, 46: 837
[4] K. E. Ranaghan, A. J. Mulholland, Int. Rev. Phys. Chem. 2010, 29: 65
[5] C. J. Woods, F. R. Manby, A. J. Mulholland, J. Chem. Phys. 2008, 128: 014109
[6] K. E. Ranaghan, L. Ridder, B. Szefczyk, W.A. Sokalski, J.C. Hermann & A.J. Mulholland, Mol. Phys. 2003, 101: 2695-2714