Why calculate wavefunctions?
· To understand and predict
the structure and reactions of molecules, we need to know about how electrons
are arranged in them.
· To study
electrons, we need to use quantum mechanics.
· In principle,
all the information we need about a molecule is contained in its wavefunction
(the wavefunction is a function of the coordinates of all the electrons and all
the nuclei).
· The trouble
is, these are difficult to calculate.
· We can make a
first simplification by concentrating on the wavefunction for the electrons
separately from that of the nuclei (the Born-Oppenheimer approximation).
· The electronic
wavefunction for a molecule will tell us all about its electronic structure -
i.e. how the electrons are arranged in the molecule.
But how do we
find the electronic wavefunction? As you have seen in Prof. Allan’s and Prof.
Balint-Kurti’s lectures, the overall molecular wavefunction is usually
approximated as a product of molecular orbitals. Each molecular orbital is then
treated as being made up of a combination of atomic orbitals (LCAO
approximation).
A useful and
quick approximate way of finding MOs for conjugated hydrocarbons uses a set of
approximations first developed in the 1930s – before computers could be used to
calculate MOs. These approximations are still useful today for quick ‘back of
the envelope’ calculations. They give important insight into the structure,
properties and reactivity of conjugated molecules.
Hückel Theory
Approximate treatment of conjugated p-electron systems.
see: Atkins &
De Paula, Physical Chemistry, 7th edn., pages 433-438.

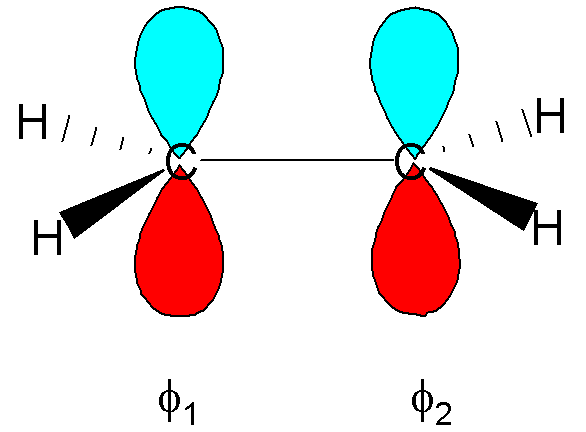
In this equation i
labels the particular MO, p the individual carbon atoms. f1 and f2 are the carbon 2p
atomic orbitals (AOs) as shown in the figure.
Note that in
this case there are only two AOs (p=1,2) and consequently only two MOs (i=1
and 2).
The cip are
the MO coefficients which are determined by minimizing the energy (i.e., using
the variation principle) - we now discuss this in detail.