 |
|

|
| |
The Configuration Interaction (CI) Method |
|||
|
The Hartree-Fock method produces an energy that is higher than the actual
value (a consequence of the variational principle), due to the approximation
of the wavefunction – the Schrödinger equation is not actually
separable, and so the molecular orbital approximation introduces inaccuracy
in this respect. It also treats coulombic repulsion between electrons
in an average way only. For a more accurate picture, the instantaneous
interaction between electrons must be considered – in the helium
atom, for example, if one electron is near the nucleus at any given point
in time, then it is energetically more favourable for the other electron
to be further away from the nucleus. i.e. The probability density of finding
another electron in the area immediately surrounding an electron is very
small. In this way, the motion of the electrons is said to be correlated,
and it is this instantaneous electron interaction (not just an average
repulsion) that is referred to as electron correlation.
Configuration interaction (CI) is a method
that includes instantaneous electron correlation. Also called configuration
mixing (CM), this involves first- and higher-order corrections
to the Hartree-Fock wavefunction that mix in elements of higher atomic
orbitals, found in excited states. The exact wavefunction is represented
as a linear combination of N-electron ‘trial’
functions, or configurations, and the linear variational method is used
to optimise the coefficients of the different configurations (see Szabo
and Ostlund[16]).
In principle, the basis set of N-electron wavefunctions used could be
complete, in which case an exact energy would be obtained. This is called
full CI. It is, however, computationally extremely expensive and
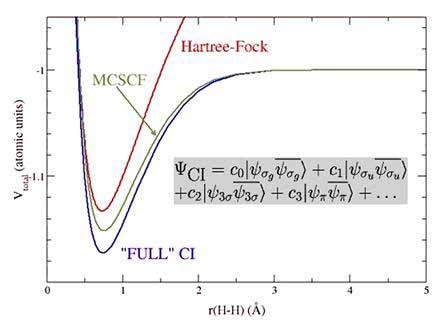
so generally the basis set is limited to a finite size. Figure 8.1 shows the potential energy curve for H2 calculated using Hartree-Fock SCF, MCSCF and full CI methods:
8.1 The Multi-Reference Configuration Interaction (MRCI) Method In conventional CI, the SCF wavefunction is used as a starting point
(called the reference function) for obtaining
the configuration state functions. |
||||
| « Previous | Next » | ||||
|
[23] G. Yan, D. Xie and A. Tian, J. Phys. Chem., 98, 8870, 1994 [24] H.-J. Werner, Adv. Chem. Phys., 69, 1, 1987 [25] H.-J. Werner and P.J. Knowles, J. Chem. Phys., 89, 5803, 1988 [26] P.J. Knowles and H.-J. Werner, Chem. Phys. Lett., 145, 514, 1988 | ||||
|
Potential Energy Surfaces and Conical Intersections • June 2002 • Ian Grant |