Molecular Mechanics
·
Even faster (‘cheaper’) calculations of the structures and energies
of molecules are possible using ‘molecular mechanics’.
·
These methods forget about electrons and nuclei, and treat atoms
as point charges with van der Waals radii.
Bonds are treated as springs (simple harmonic energy terms), with simple
terms also for torsion angles and bond angles.
·
These functions contain parameters (bond lengths, force constants,
etc.) which are optimized to reproduce the structures of molecules as found
experimentally or by ab initio calculations.
EMM = Ebond + Eangle + Etorsion
+ EVDW + Eelectrostatic
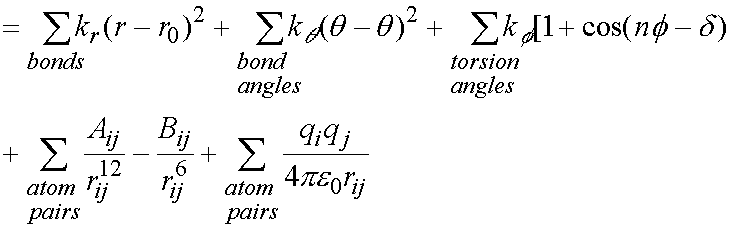
(32)
·
With molecular mechanics (MM) methods, it
is possible to study very large molecules (solids, polymers, surfaces,
proteins, DNA, etc.).
·
The motion of such molecules can be
studied using MM energy functions in molecular dynamics simulations.
·
One application is to study protein
folding – e.g. the IBM ‘Blue Gene’ project.
However,
MM potential functions are not suitable for modelling chemical reactions. The functional form is incorrect (e.g. a
harmonic term for stretching a bond does not allow the bond to break); also the
use of stable molecules in the parameterization process means that only stable
molecules can be described by these functions, and not e.g. transition
states. MM methods are described more
fully below.