·
All the thermodynamic properties of
the molecule can now be calculated.
·
Usually we are interested in
calculating energy differences, not absolute energies.
·
Free energy differences give
equilibrium constants and rate constants.
·
As you have seen in earlier courses, The
equilibrium constant, K, for a
reaction is related to the standard Gibbs free energy for reaction, DrGq :
·
DrGq = –RT ln K
· Calculations are more useful for studying rate constants than equilibrium
constants (equilibrium constants can usually be measured straightforwardly
experimentally or derived from tables of thermodynamic data – rate constants on
the other hand depend on the properties of the transition state, and transition
states are not easily studied by experiment).
· From transition state theory, rate constants may be expressed in
terms of the activation free energy, D‡G, (the free
energy difference between reactants and the transition state), e.g. for a
first-order reaction:
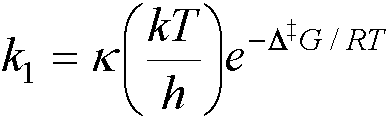
A standard activation enthalpy, D‡H, and standard
activation entropy, D‡S, can be
defined:
D‡G = D‡H – TD‡S
·
We have
assumed that the total energy can be written as a sum of different terms (translational,
rotational, vibrational and electronic).
This is a good approximation.
·
In
exactly the same way, the enthalpy and entropy of a molecule can be written as
a sum of contributions:
H = Htrans + Hrot + Hvib
+ Helec
S = Strans + Srot + Svib
+ Selec
By
calculating each of these terms for the reactant(s) and transition state of a
reaction, we can calculate the activation free energy as D‡G = D‡H – TD‡S.
For one mole of a (non-linear) molecule in the gas
phase, Htrans =5RT/2, Hrot= 3RT/2.
The vibrational enthalpy is given by:
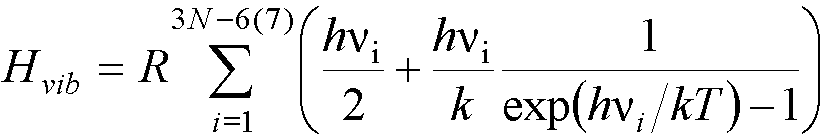
where k is again the Boltzmann
constant.
·
You don’t need to be able to remember
this equation. You do need to notice (and be able to
explain) a couple of things about it.
·
First, the sum is
over 3N-6 normal modes for a stable
(non-linear) molecule, but over 3N-7
modes for a transition state.
·
There is one less normal mode for a
transition state because the motion along the reaction coordinate is not
included as a normal vibration.
·
Remember that in a normal mode analysis
of a transition state, one of the modes has an imaginary frequency –this is the
reaction coordinate, and is not included in the sum.
·
The other thing to notice (and
remember) about this expression is that it is made up of two parts, the first
is the zero-point energy, and the second depends on the temperature. This is
the contribution to the enthalpy from molecules that are not in the vibrational
ground state.
There are
similar expressions (again derived from the partition function) for the
entropy.
The
difference in enthalpy and entropy between the reactant(s) and transition
states can them be calculated (e.g. for a bimolecular
reaction, D‡Htrans = -5/2RT (going from two moles of
molecules (reactants) to one mole (transition state), similarly D‡Hrot = -3/2RT).
Adding the
enthalpy and entropy differences between the reactant(s) and the transition
state gives the activation free energy D‡G = D‡H – TD‡S.
Measuring relative
to the energy of the reactants, the enthalpy of the reactants is zero, and that
of the TS is DE‡, the
difference between the electronic energy of the reactants and the transition
state
(e.g. given by an ab initio calculation – remember that transition states
cannot be modelled by standard molecular mechanics. Remember also that a method
that includes
electron correlation would be needed to calculate the energy of the
transition state accurately).