Calculating
activation free energies - summary
·
To
summarize, to calculate a rate constant, we need to calculate D‡G.
·
This can be done if we know the
structure, energy and normal modes of the molecule.
·
Remember there are approximations
involved – e.g. treating all vibrations as harmonic, which as you know is a
poor approximation for low energy, ‘floppy’ motions, and for internal
rotations. The most important contribution to the barrier is usually the
electronic energy.
·
Barriers can be calculated to within
approximately 0.1 kcal/mol only for very simple systems, and it is only at this
level of accuracy that it is usually worth going beyond the harmonic oscillator
approximation. It is difficult to calculate activation energies to within 1
kcal/mol.
·
Even a calculation of D‡G accurate to within 0.2
kcal/mol would give an error of a factor of 1.4 at 300K in a rate constant
(recall the expression for a first-order rate constant above). This is worse
than can be achieved by experiment for these simple systems.
· Calculations are more useful for understanding and predicting relative
rates, e.g. predicting the effects of substituents, ratios between different
possible products. They help with the interpretation of experiments. Also, calculations allow transition states and reactive intermediates
to be studied directly (this is difficult by experiment, particularly for large
molecules, and in condensed phases)
Solvent Effects
·
The
examples above are for small molecules in the gas phase.
·
It is
possible to include the effects of solvent in approximate ways (i.e. not
including solvent molecules explicitly in a calculation).
·
For
example, in a quantum chemical calculation (e.g. ab initio, semiempirical or
density-functional), solvation energy can be calculated by treating the solvent
as a uniform medium with a dielectric
constant e.
·
The molecule
occupies a cavity in the solvent (the simplest models use a sphere big enuogh
to encompass the molecule, but better models use a cavity based on e.g the van der
Waals radii of the atoms in the molecule, to define a cavity based on the
molecular shape).
·
A
molecule’s solvation free energy can then be written as the sum of different
terms:
DGsolvation = DGcavity + DGdispersion + DGelectrostatic
Creating the cavity within the solvent costs energy (DGcavity),
while there are favourable
dispersion interactions
between the solvent and the solute (DGdispersion). These two terms are usually combined, and assumed to be proportional
to the solvent-accessible surface area of the molecule. Different atom types
are different parameters to relate the solvation energy to their
solvent-exposed surface area.
The simplest estimate of the electrostatic component of the solvation
energy (DGelectrostatic) is given by the Born model, which treats the cavity as a sphere for a molecule with charge q. The Onsager model is
similar (a molecule is represented simply by its dipole). Better models include
more complete representations of charge distribution in the molecule (e.g. based
on the electrostatic potential), and also polarization due to the solvent.
· These methods are used in ab initio
calculations because the computational demands of high-level calculations
prevent (many) solvent molecules being included explicitly.
In molecular
mechanics, one
effect of solvation (the screening of charges) can be included approximately by
altering the dielectric constant used in the calculation of electrostatic
interactions – this reduces
the interaction energy between charges qi and qj by the factor 1/e where e is the solvent dielectric constant.
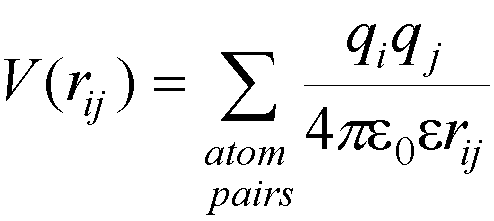
Alternatively, the dielectric constant can be made distant dependent (making the dielectric constant proportional
to r), reducing the interaction energy two charges
by 1/r where r is the distance between them, i.e.
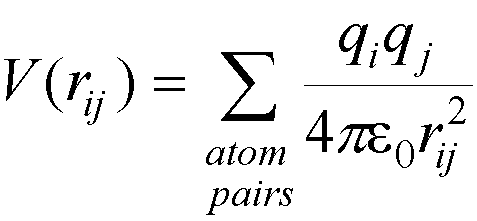
greatly reducing the interaction between different groups.
·
However, these are highly approximate approaches.
·
There are better ‘implicit solvent’ models for molecular mechanics,
which can give good results, and allow large molecules to be treated in long
simulations, while including solvation effects.
·
Ideally though, we want
to include solvent molecules explicitly.