Molecular
Simulation – continued
Leach, Chapter 6 (and 7 & 8);
Grant and Richards Chapter 4
Monte Carlo (MC) and Molecular Dynamics (MD)
These are the two most common simulation methods in molecular
modelling.
Boundary conditions
· Even with
approximate methods, cannot simulate very large numbers of atoms.
· Get round this
by using periodic boundary conditions (as shown previously for lattice
dynamics) – the simulation system is replicated in all directions, and interacts
with its ‘images’, giving a pseudo-infinite system.
· Alternatively,
can focus on just a small region of a large molecule, and using some boundary
restraints at the edges to make sure the structure is maintained, and the
effects of the environment are included approximately.
Monte
Carlo
To calculate a particular property, Q, of a system with a
constant number of particles, temperature and volume (the NVT or canonical
ensemble): statistical mechanics shows that the average value of Q, <Q>
is given by:
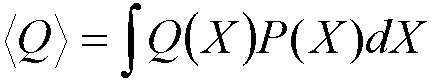
P(X) is the Boltzmann weighted probability:
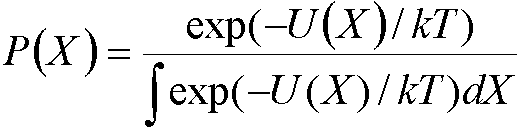
where U(X) is the internal energy (e.g. calculated
by molecular mechanics) and X represents all possible configurations of the
system.
·
You might have thought the values of these integrals could be
found by simply sampling different configurations of the system by moving atoms
at random, calculating the value of Q(X) at each configuration,
and averaging to get <Q>.
·
However, this wouldn’t work – each configuration would make the
same contribution, whereas in fact the probability term means that only
configurations with low energies make a significant contribution to P(X).
·
The correct way to do the sampling of configurations is to use
the Metropolis method: in this the generation of configurations is
biased towards those that make the most important contributions.
·
Specifically, configurations are generated with a Boltzmann
probability exp(-U(X)/kT.
·
So the Monte Carlo method generates configurations randomly and
uses a specific set of criteria (usually the Metropolis method) to decide
whether or not to accept each new configuration.
·
These criteria ensure that the probability of obtaining a given
configuration is equal to its Boltzmann factor exp (-U(X)/kT).
·
U(X) would be calculated by molecular mechanics,
for example.
·
Configurations with low energy are generated with higher
probability than configurations with higher energy.
For each configuration that is
accepted, the values of the desired properties are calculated, and at the end
of the calculation, the average of these properties is obtained by simply
averaging over the number of configurations, M:
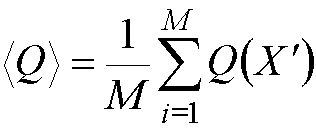
where X' indicates that only
configurations with an acceptable Boltzmann weighted probability have been
sampled. (Q could be for example the internal energy, calculated as the
average of the energies of the configurations generated in the simulation).
In a Monte Carlo simulation, each new configuration of the
system is generated by a random move of an atom or molecule, or (in the case of
large molecules such as polymers or proteins) by more sophisticated ‘moves’,
such as rotation around bonds or concerted motions. The energy, U(X), is
calculated for the new configuration.
Then
·
If the energy of the new configuration is lower than
the energy of its predecessor, the new configuration is accepted.
·
If the energy of the new configuration is higher
than the energy of its predecessor, then the Boltzmann factor of the
energy difference is calculated: exp (-DU/kT). The
Boltzmann factor is compared with a random number between 0 and 1. If the
random number is lower than the Boltzmann factor, the new configuration is
accepted and the configuration is included. If the random number is higher,
then the new configuration is rejected, and the original configuration is used
for the next iteration
·
This procedure allows moves to states of higher energy. The
smaller the increase in energy (DU), the greater
the probability that the configuration will be accepted.
Monte Carlo simulations are used for simulations of liquids,
solids, solutions, polymers, proteins, etc.
Next: molecular dynamics simulations